
Abstract
Xylanases (EC 3.2.1.8) have been considered as a potential green solution for the sustainable development of a wide range of industries including pulp and paper, food and beverages, animal feed, pharmaceuticals, and biofuels because they are the key enzymes that degrade the xylosidic linkages of xylan, the major component of the second most abundant raw material worldwide. Therefore, there is a critical need for the industrialized xylanases which must have high specific activity, be tolerant to organic solvent or detergent and be active during a wide range of conditions, such as high temperature and pH. In this study, an extracellular xylanase was purified from the culture broth of Aspergillus niger VTCC 017 for primary structure determination and properties characterization. The successive steps of purification comprised centrifugation, Sephadex G-100 filtration, and DEAE-Sephadex chromatography. The purified xylanase (specific activity reached 6596.79 UI/mg protein) was a monomer with a molecular weight of 37 kDa estimating from SDS electrophoresis. The results of LC/MS suggested that the purified protein is indeed an endo-1,4-β-d-xylanase. The purified xylanase showed the optimal temperature of 55 °C, and pH 6.5 with a stable xylanolytic activity within the temperature range of 45–50 °C, and within the pH range of 5.0–8.0. Most divalent metal cations including Zn2+, Fe2+, Mg2+, Cu2+, Mn2+ showed some inhibition of xylanase activity while the monovalent metal cations such as K+ and Ag+ exhibited slight stimulating effects on the enzyme activity. The introduction of 10–30% different organic solvents (n-butanol, acetone, isopropanol) and several detergents (Triton X-100, Tween 20, and SDS) slightly reduced the enzyme activity. Moreover, the purified xylanase seemed to be tolerant to methanol and ethanol and was even stimulated by Tween 80. Overall, with these distinctive properties, the putative xylanase could be a successful candidate for numerous industrial uses.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In our age, the exponential economic development has been achieved at the cost of severe overexploitation of natural resources. The green industrial transformation that refers to processes within industries to reduce environmental change impact is an essential requirement for balancing environmental sustainability and positive economic development [1]. This green transformation needs green technologies that economize on exhaustible resources and emit fewer pollutant discharge [2]. Thanks to the nontoxic and eco-friendly characteristics, stability and catalytic activity, microbial enzymes have gained interest for their ability to replace conventional toxic agents in a wide range of industries [3]. Along with cellulase and pectinase, xylanase is the third industrially prevalent enzyme [4]. The diversified application of xylanase covers several industrial fields including food, paper, textile, pharmaceutical, and biorefinery. Among them, two domains in which xylanase has been used widely in the industry are paper and food. In the paper processing, xylanase has been used to substitute harmful bleaching agents like chlorine [5]. In the food industry, this enzyme has been used as an additive during the bread-making and fruit-juice clarifying process [6, 7]. Besides, xylanases take a major role in the increase of the digestibility of xylan containing-food, and reduce the viscosity in the animal gastrointestinal tract leading to increase food absorption. This led to improving beneficial intestinal microbiota and reduce digestive disorders. Xylanase also helps to maximize feed efficiency and therefore save costs and improve productivity [8]. That why the demand for xylanase, recently, has been growing tremendously in the biotechnology industry. As a result, xylanase is required in large quantities in each circumstance with specific properties. A solution to this demand is studying the enzyme from various sources. Xylanases can be found abundant in nature (e.g., mollusks, insects, and microorganisms), and in recent years, many kinds of xylanases have been isolated from various microorganisms including bacteria, actinomyces, yeasts, molds, and algae [9]. Among of them, Aspergillus sp. are potential producers for xylanase [10, 11]. The enzymes produced have higher activity than other eukaryote hosts and have some outstanding features such as specific structural regions which we can use mutation techniques, recombinant proteins to significantly increase catalytic efficiency and thermostability [11,12,13]. Therefore, the purpose of this study is to purify and characterize A. niger VTCC 017 xylanase after optimization of culture conditions (Table 1).
Materials and Methods
Chemicals and Reagents
Birchwood xylan, 3,5-dinitrosalicylic acid (DNS), SDS were from Sigma-Aldrich (USA), Sephadex G100, DEAE-Sephadex, and anion exchange column were purchased from Pharmacia Co; Tween 20 and Tween 80 were purchased from BioBasic Inc. (USA); Triton X-100 was purchased from Merck, Germany. All other chemicals were of analytical grade unless otherwise stated.
Strain and Culture Conditions
The strain A. niger VTCC 017 was obtained from Vietnam Type Culture Collection (VTCC). It was inoculated in the liquid medium containing (g/L): corncob 35, soybean powder 5, and pH 5.0. The inoculated flasks were incubated for 144 h at 30 °C on a rotary shaker at 180 rpm.
Xylanase Purification
The broth was centrifuged at 11,180 rcf for 10 min. A 5 ml of the extracellular supernatant (195.43 IU/mL) was loaded to a Sephadex G-100 column (2.6 × 6 cm) at a flow rate of 24 mL/h pre-equilibrating with 50 mM potassium phosphate buffer pH 7.5. The eluate was collected with 2 mL per fraction. A highly active xylanase pool of 4 mL through Sephadex G-100 column was loaded onto an anion exchange column (DEAE-Sephadex, 2.6 × 26 cm) pre-equilibrated with 50 mM Tris HCl buffer pH 8.0 containing 50 mM NaCl, then washed with the same buffer. The protein was eluted with 50 mM Tris HCl buffer pH 8.0 containing 1 M NaCl at a flow rate of 24 mL/h. The eluate was obtained with 2 mL per fraction. The fractions containing strong, potent xylanase activity were pooled and used as purified enzymes for characterization. All purification steps were carried out at 4 °C, unless otherwise specified.
Xylanase Activity Assay
Xylanase activity was determined based on Baileys et al. (1992) methodology [14] and reducing sugar was measured using the 3,5-dinitrosalicylic acid [15]. Briefly, a mixture of 100 µL of the crude or purified xylanase containing 0.06 µg total protein was incubated with 400 µL of 0.5% (w/v) birchwood xylan in 20 mM potassium phosphate buffer pH 6.5 at 55 °C for 5 min. The reaction was terminated by adding 1.25 mL of DNS reagent. The absorbance of the mixture was then assessed at a wavelength of 540 nm. D-xylose was used as standard. One unit (IU) of xylanase activity was defined as the amount of xylanase needed to produce 1 μmole of xylose equivalent per minute under the above assay conditions.
SDS-PAGE and Protein Concentration Determination
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was performed using a 12.5% (w/v) polyacrylamide gel according to the description in Laemmli’s research [16]. Protein molecular weight marker (Cat. # SM0661, Fermentas) was used as a molecular weight marker. The proteins on the gel were visualized by staining with Coomassie Brilliant Blue R-250 according to the manufacturer’s instructions.
Protein concentration was determined by the Bradford method using bovine serum albumin (BSA) as the standard [17]. A 100 μL of xylanase-containing sample was mixed with 1000 μL of Bradford reagent, followed by incubation for 2 min at room temperature. The absorbance was measured at a wavelength of 595 nm against the reagent blank. The total protein concentration was determined by referring to the standard curve constructed using bovine serum albumin (BSA) as standard.
Protein Identification
The purified xylanase was identified by liquid chromatography–mass spectrometry (LC/MS) assay. Peptides were extracted from trypsin-digested protein samples using the standard method [18]. Peptides were analyzed by electrospray ionization mass spectrometry using the Shimadzu Prominence nano HPLC system (Shimadzu) coupled to a 5600 TripleTOF mass spectrometer (Sciex). Tryptic peptides were loaded onto an Agilent Zorbax 300SB-C18, 3.5 µm (Agilent Technologies) and separated with a linear gradient of water/acetonitrile/0.1% formic acid (v/v). Spectra were analyzed to confirm protein identity using Mascot sequence matching software (Matrix Science) with UniProt database. Peptide fragments showing individual ions scores > 55 were identified uniquely or extensive homology (p < 0.05).
Characterization of Purified Xylanase
The Optimal Temperature and Thermal Stability
The optimal temperature of the purified xylanase activity was determined in the range of 40–80 °C by measuring enzyme activity of 100 µL of the purified enzyme (0.06 µg protein) in 20 mM potassium phosphate buffer pH 6.5 for 5 min.
Thermal stability of the purified xylanase was determined by assessing the residual enzyme activity under standard conditions (100 µL of the purified enzyme (0.06 µg protein), pH 6.5, 55 °C, and 5 min) after incubation of the enzyme at either 37, 40, 45, 50, 55, or 60 °C for different intervals of time. The initial xylanase activity measured at 55 °C was considered 100% activity; the residual activity was displayed as the ratio of the initial activity.
The Optimal pH and pH Stability
The optimal pH for xylanase activity of was determined in pH ranging 3.0–8.0 (acetate buffer: 3–5 and phosphate buffer: 6–8) by assessing the activity of 100 µL of the purified enzyme (0.06 µg protein) at 55 °C for 5 min.
The pH stability of purified xylanase was determined by measuring residual enzymatic activity under standard conditions (100 µL of the purified enzyme (0.06 µg protein), pH 6.5, 55 °C, and 5 min) after pre-incubating the enzyme at 30 °C in the buffers mentioned above for 1–8 h. The initial xylanase activity measured at pH 6.5 was considered 100% activity; the residual activity was displayed as the ratio of the initial activity.
Effect of Metal Ions on Xylanase Activity
To investigate the effects of different metal ions on the activity of the purified xylanase, the enzyme was incubated in 20 mM Tris buffer pH 6.5 containing 10 mM of various metal ions (Mg2+, Fe2+, Cu2+, Ca2+, Ni2+, Mn2+, Zn2+, Ag+, K+, and EDTA) at 30 °C for 60 min. Incubation of purified xylanase in the absence of added reagents was the control experiment. The residual activity was measured under standard assay conditions (100 µL of the purified enzyme (0.06 µg protein), pH 6.5, 55 °C, and 5 min). The residual activity was displayed as the ratio of the control activity.
Effect of Organic Solvents and Detergents on Xylanase Activity
We investigate the effects of different organic solvents and detergents on the activity of purified xylanase by incubating the enzyme in 20 mM potassium phosphate buffer pH 6.5 containing 10–30% of various solvents (methanol, ethanol, isopropanol, acetone, and n-butanol) and 2% of various detergents (SDS, Tween 20, Tween 80, and Triton X-100) at 30 °C for 60 min. Incubation of purified xylanase in the absence of added reagents was the control experiment. The residual activity was measured under standard assay conditions (100 µL of the purified enzyme (0.06 µg protein), pH 6.5, 55 °C, and 5 min). The residual activity was displayed as the ratio of the control activity.
Kinetics Study
The kinetic parameters such as maximum reaction velocity (Vmax) and Michaelis constant (Km) were estimated by calculating activity of the purified xylanase against Birchwood xylan. The xylanase assay was performed at 55 °C, keeping enzyme concentration constant (0.06 µg protein), against different substrate concentration ranging from 1 to 10 mg/mL. Both Vmax and Km were calculated from Michaelis–Menten equation using nonlinear adjustment and Lineweaver–Burk double reciprocal plots.
Statistical Analysis
All measurements were carried out in triplicate and data were expressed as mean with error bars as standard deviation.
Results
Purification of A. niger VTCC 017 Xylanase
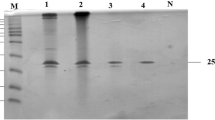
Following 144 h of cultivation under optimized culture conditions [19], A. niger VTCC 017 exhibited the highest xylanase activity at 195.43 IU/ml (specific activity of 1931.27 IU/mg protein). This culture supernatant was loaded to Sephadex G-100 and the xylanase activity of collected fractions was assayed (Fig. 1A). Peak fractions with high enzymatic activities were subjected to SDS-PAGE to determine the purity (Fig. 1B). The pooled Sephadex G-100 fractions containing high xylanase activity (1866.64–2665.6 IU/mg) were applied further to the DEAE-Sephadex anion exchange chromatography (Fig. 2). The purified enzyme revealed a single band on SDS-PAGE with a molecular mass of 37 kDa (Fig. 2B).

Sephadex G-100 chromatography of the xylanase from A. niger VTCC 017 (A) (square: protein concentration (µg/ml), circle: xylanase activity (IU/ml)), column equilibrated and eluted with 50 mM potassium phosphate buffer pH 7.5; flow rate 24 mL/h, fraction size 24 mL, and SDS-PAGE of high-activity fractions (B) (1: the crude supernatant; 2–8: fractions from 3- 9; M: maker)

DEAE-Sephadex anion exchange chromatography of the xylanase from A. niger VTCC 017 (A) (square: protein concentration (µg/ml), circle: xylanase activity (IU/ml)) and SDS-PAGE of high-activity fractions (B) (1: the crude supernatant; 2: fractions after through Sephadex G-100 column; 3–8: fractions from 12 to 14, 16, and 17 after through DEAE-Sephadex column; M: maker)
Protein Identification
The single protein on SDS-PAGE (Fig. 2B) was cut out from the gel and used for LC/MS analysis. The sequences of these peptide fragments were KYLGNIGDQYTLTK (position 41–53), ADFGALTPENSMK (position 63–76), GQFSFSGSDYLVNFAQSNNK (position 85–103), DSVFYKVIGEDYVR (position 166–179), LYINDYNLDSASYPK (position 195–209) (Fig. 3). The results of a BLAST search indicated that these amino acid sequences were highly homologous (ion scores above 55, p < 0.05) with an endo-1,4-beta-xylanase of the glycoside hydrolase family 11 from A. niger (AIC36735.1) and A. niger CICC 2475 (AFZ94943.1) (Fig. 4).

Mascot search results of five neutral identified peptides A KYLGNIGDQYTLTK, B ADFGALTPENSMK, C GQFSFSGSDYLVNFAQSNNK, D DSVFYKVIGEDYVR, and E LYINDYNLDSASYPK found in Genbank AOAOS2WJ5 xylanase from (GenBank, A0A060IU57) corresponding to ion scores of with p < 0.05, respectively

Peptide sequence alignment of fives neutral identified peptides (5 peptides) with endo-1,4-beta-xylanase from A. niger (AIC36735.1) and endo-1,4-beta-xylanase from A. niger CICC 2475 (AFZ94943.1)
The Optimal Temperature and Thermal Stability
The effects of temperature on the activity of purified enzymes are shown in Fig. 5. The xylanase activity increased gradually from 57.8% (1590.4 IU/mg) at 40 °C to the maximum of 100% (2749.3 U/mg) at 55 °C (Fig. 5A) and then decreased gradually to 41.1% (1128.8 U/mg) at 80 °C. This result indicates that the optimal temperature of the purified xylanase was 55 °C at pH 6.5.

Effect of temperature on the enzyme activity (A) and the thermal stability (B) of xylanase purified from A. niger VTCC 017
The thermal stability was determined by pre-incubating the enzyme at either 37, 40, 45, 50, 55, or 60 °C for 8 h. Samples were taken after every one hour. The results (Fig. 5B) showed that’s the purified xylanase from A. niger VTCC 017 was stable after incubation at 45 °C and 50 °C. The residual xylanase activity retained 85%, and 95% for 8 h at 50 °C, and 45 °C, respectively. At other tested temperatures, the residual activity was reduced significantly to 50% after 2 or 4 h of incubation.
The Optimal pH and pH Stability
The effects of pH on the activity of purified enzymes are shown in Fig. 6. The purified xylanase from A. niger VTCC 017 worked in a relatively broad range of pH 3.0–8.0. The xylanase activity increased gradually from 59.3% (3636 U/mg) at pH 3.0 to the maximum of 100% (6128 IU/mg) at pH 6.5 and then decreased gradually to 76.4% (4679 IU/mg) at pH 8.0 (Fig. 6A). This result indicates that the optimal pH of purified xylanase was 6.5.

Effect of pH on the enzyme activity (A) and pH stabitity (B) of the xylanase purified from A. niger VTCC 017
A similar pH profile was seen when the purified xylanase was treated at pH 3.0–8.0 for 8 h (Fig. 6B). The enzyme was stable over a relatively broad pH range, retaining more than 80% of the maximum activity after 8 h of incubation at pH 5, 6.5, 7, and 8. These results suggested that the purified xylanase might be an alkaline stable enzyme.
Effect of Metal Ions on Xylanase Activity
The activity of the purified xylanase in the presence of different metal ions is shown in Table 2. The enzyme activity was reduced to 50% by Zn2+ at the concentrations of 10 mM. Slight inhibition was observed in the presence of some metal ions at 10 mM concentration, such as Fe2+, Cu2+, Mg2+, and Mn2+. In contrast, the Ag+, Ni+, K+, and EDTA partially enhanced the activity of the purified xylanase.
Effect of Organic Solvents and Detergents on Xylanase Activity
Organic solvents and detergents resistance are another required characteristic of an industrial enzyme. The effects of different organic solvents and detergents on xylanase activity are shown in Fig. 7 and Table 3, respectively.

Stability of the xylanase purified from A. niger VTCC 017 in the presence of various organic solvents. (MeOH: methanol, EtOH: ethanol, IsOH: isopropanol, and n-BtOH n-butanol)
Organic solvents like methanol, ethanol acetone, isopropanol, and n-butanol were used to evaluate xylanase activity. A partial decrease in enzyme activity was found in the presence of 10–30% (v/v) acetone, isopropanol, and n-butanol. Maximum stability was observed in presence of methanol followed by ethanol. The residual xylanase activity retained more than 60% in the presence of 30% (v/v) methanol or ethanol (Fig. 7).
Among the detergents tested, SDS at 2% (w/v) significantly inhibited the enzyme activity while Triton X-100 and Tween 20 had little effect on the activity. In contrast, Tween 80 slightly increased the activity of the purified xylanase (Table 3). These results are similar to those described for many Aspergillus sp. such as A. japonicus [20], A. fumigatus Z5 [21].
Kinetics Study
The maximum velocity (Vmax) and Michaelis–Menten constant (Km) of the purified enzyme using Birchwood xylan as a substrate were calculated using Lineweaver–Burk plot. The value of Vmax and Km were estimated to be 2000 U/mg and 1.8 (mg/mL), respectively (Fig. 8). This result showed that the purified enzyme has relatively high affinity for Birchwood xylan substrate.

Lineweaver–Burk plot for the xylanase purified from A. niger VTCC 017 using Birchwood xylan as the substrate
Discussion
Xylanase has attracted increasing attention in recent years due to its efficient application in various industrial sector. Efforts have been made to produce this potential enzyme at large scale to fulfill the market demand. In order to achieve this purpose, the screening for novel potential industrialized xylanase from natural sources should be conducted parallel with the development of engineering technologies to improve catalytic characteristics of natural xylanses as well as the enzyme production efficiency. The present study focuses on purification, identification, and characterization of an extracellular xylanase from a local fungus, Aspergillus niger VTCC 017. To purify xylanase from fungi, there are a variety of procedures, but generally, there is a combination of gel filtration chromatography and ion-exchange chromatography. In this study, after two-step chromatography, the specific activity of purified xylanase was 6596.79 IU/mg with a final yield of 28.5%, and a purification factor of 3.42. These results are encouraging when compared to several similar studies [22, 23]. The molecular weight of the purified enzyme were estimated to be 37 kDa after SDS-PAGE analysis. This molecular weight of the purified xylanase was close to the molecular weight of most of the purified xylanases from Aspergillus sp. strains which have a molecular weight of about 21–35 kDa such as, 21 kDa xylanase purified from A. oryzae [24] and from Aspergillus cf. niger [25], 22 kDa xylanase was produced by a Mexican Aspergillus sp. FP-470 strain [26], two proteins with a molecular weight of 21 kDa and 24 kDa from A. giganteus [27], 35 kDa xylanase purified from A. ficuum AF-98 [28], 34 kDa xylanase purified from A. nidulans [29], and 33.671 kDa xylanase purified from A. oryzae HML366 [11]. The protein identification results not only confirmed the origin of the purified xylanase from Aspergillus sp but also revealed that it belongs to the glycoside hydrolase family 11 (GH 11). This is such a promising result because compared to other xylanases, GH11 xylanases display several interesting properties such as high substrate selectivity and high catalytic efficiency, small size, variety of pH, and temperature optimum, making them suitable for various conditions in many applications [30]. To evaluate the potential application of the purified xylanase, in the second part of the present study, the biochemical and catalytic characteristic of this enzyme were investigated. The first measured parameters were the optimal temperature and the thermal stability. As most of the industrial processes are generally carried out at high temperature, the thermostable enzymes have many advantages over mesophyllic enzymes. The results showed that, the purified xylanase might be considered as a thermostable enzyme since it was active in a large temperature range from 40 to 80 °C and showed its optimal temperature at 55 °C at pH 6.5. This optimal temperature is similar to those from various Aspergillus strains which range from 45 to 60 °C [31]. Many factors might affect the thermostability of enzymes such as the structure of N-terminal region, sequence of C-terminus, presence of disulfide bonds, percentage of hydrophobic aminoacid residues, etc. [32, 33]. However, according to Gabriel Paës et al. (2012) [30], the thermostability of members of GH 1 xylanase family like our purified enzyme might originate from different features, none of them being absolutely required. Thermostability or thermoactivity factors seem unique to a given enzyme and are not universal. In our next step, a detailed 3D structural analysis will be performed to reveal the key elements contributing to the thermal-characteristic of the purified xylanase. Like for the optimal temperature and the thermal stability, the pH optimal of the purified xylanase are also important parameters for an industrial enzyme. Currently, a major application of xylanase is in pulp and paper industries where xylanases work under the alkaline conditions. The purified xylanase from Aspergillus niger VTCC 017 had optimum pH of 6.5 and was stable over a relative broad pH range, retaining more than 80% of the maximum activity after 8 h of incubation at pH 5, 6.5, 7, 8. This is a significant characteristic because most fungal GH11 xylanases are acidophilic [30]. To our knowledge, only a few xylanases have the stability under alkaline condition as the purified xylanase did; these include xylanases from Aspergillus tamarii kita [34] and Aspergillus flavus [35]. The mechanism of high pH adaptation of xylanase is due to a highly acidic, negatively charged surface, and a deeper active site cleft [36]. Therefore, further studies on crystallization and site-directed mutagenesis are needed to clarify the mechanisms causing our purified xylanase stable under alkaline conditions.
When challenged with other industrial hazardous factors like various organic solvents and detergents, the purified xylanase also gained positive results. The enzyme was stable in the presence of conventional organic solvents like methanol, ethanol, and isopropanol. Especially, the residual xylanase activity retained more than 60% in the presence of 30% (v/v) methanol or ethanol. To date, few organic solvent-tolerant xylanases have been purified and characterized [37,38,39,40]. Therefore, these results suggested that the purified xylanase might become a potential candidate for various industrial applications.
The previous studies showed that metal ions have different effects on the activity of different xylanases from different Aspergilus sp. Cu2+, for example, strongly inhibited the xylanase from A. giganteus [27] but slightly enhanced the xylanase from A. ficuum AF-98 [28]. Zn2+ enhanced the xylanase activity from A. cf. niger BCC14405 [25] and from A. sydowii SBS 45 [23] but inhibited the xylanase from A. giganteus [27]. These results indicate the diversity of structure of xylanase’s active sites. In our study, the activity of the enzyme was significantly inhibited by Zn2+. This result suggested that the purified xylanase might contain Cys and His in its active site [41]. Further investigations are needed to confirm this attractive hypothesis.
Conclusion
In this study, a GH 11-xylanase from A. niger VTCC 017 has been successfully purified and biochemically characterized. The purified xylanase had high specific activity (6596.79 IU/mg protein) and displayed good stability at alkaline pH range, high temperature range, and in the presence of conventional organic solvents like methanol and ethanol. Although the molecular mechanisms of the significant characteristics of the purified xylanase are needed to be investigated by further experiment, which is planned for the future, these encouraging first results might pave the way for a new potential candidate for various industrial applications, especially in pulp and paper industries. A novel industrial potential GH11 xylanase was successfully purified, identified, and biochemically characterized from a local filamentous fungus, A. niger VTCC 017.
References
Alfredsson, E. (2014). Prerequisites for a green structural transformation of swedish industry—Synthesis report. Growth analysis, Swedish agency for growth policy analysis.
Rodrik, D. (2014). Green industrial policy. Oxford Review of Economic Policy, 30(3), 469–491.
Singh, R., Kumar, M., Mittal, A., & Mehta, P. K. (2016). Microbial enzymes: industrial progress in 21st century. 3 Biotech, 6(2), 174.
Bajaj, P., & Mahajan, R. (2019). Cellulase and xylanase synergism in industrial biotechnology. Applied Microbiology and Biotechnology, 103, 8711–8724.
Sridevi, A., Ramanjaneyulu, G., & Suvarnalatha, D. P. (2017). Biobleaching of paper pulp with xylanase produced by Trichoderma asperellum. 3 Biotech, 7, 266.
Adiguzel, G., Faiz, O., Sisecioglu, M., Sari, B., Baltaci, O., Akbulut, S., Genc, B., & Adiguzel, A. (2019). A novel endo-β-1,4-xylanase from Pediococcus acidilactici GC25; purifcation, characterization and application in clarifcation of fruit juices. International Journal of Biological Macromolecules, 129, 571–578.
Queiroz-Brito-Cunha, C. C., Gama, A. R., Cintra, L. C., Bataus, L. A. M., & Ulhoa, C. J. (2018). Improvement of bread making quality by supplementation with a recombinant xylanase produced by Pichia pastoris. PLoS ONE, 13, 1–14.
Bedford, M. R. (2018). The evolution and application of enzymes in the animal feed industry: The role of data interpretation. British Poultry Science, 5(59), 486–493.
Bhardwaj, N., Kumar, B., & Verma, P. (2019). A detailed overview of xylanases: An emerging biomolecule for current and future prospective. Bioresources and Bioprocessing, 6, 40.
Guimaraes, N. C. A., Sorgatto, M., Peixoto-Nogueira, S. C., Betini, J. H. A., Zanoelo, F. F., Marques, M. R., Polizeli, M. T., & Giannesi, G. C. (2013). Bioprocess and biotechnology: Effect of xylanase from Aspergillus niger and Aspergillus flavus on pulp biobleaching and enzyme production using agroindustrial residues as substract. Springer Plus, 2, 380.
He, H., Qin, Y., Li, N., Chen, G., & Liang, Z. (2015). Purification and characterization of a thermostable hypothetical xylanase from Aspergillus oryzae HML366. Applied Biochemistry and Biotechnology, 175, 3148–3161.
Li, Y., Zhang, B., Chen, X., Chen, Y., & Cao, Y. (2009). Improvement of Aspergillus sulphureus endo-beta-1,4-xylanase expression in Pichia pastoris by codon optimization and analysis of the enzymic characterization. Applied Biochemistry and Biotechnology, 160, 1321–1331.
Yang, W., Yang, Y., Zhang, L., Xu, H., Guo, X., Yang, X., Dong, B., & Cao, Y. (2017). Improved thermostability of an acidic xylanase from Aspergillus sulphureus by combined disulphide bridge introduction and proline residue substitution. Scientific Reports, 7, 1587.
Bailey, M. J., Biely, P., & Poutanen, K. (1992). Interlaboratory testing of methods for assay of xylanase activity. Journal of Biotechnology, 23(3), 257–270.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugars. Analytical Chemistry, 31, 426–428.
Laemmli, U. K. (1970). Cleavage of structure proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Bradford, M. N. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Bringans, S., Eriksen, S., Kendrick, T., Gopalakrishnakone, P., Livk, A., Lock, R., & Lipscombe, R. (2008). Proteomic analysis of the venom of Heterometrus longimanus (Asian black scorpion). Proteomics, 8, 1081–1096.
Do, T. T., Quyen, D. T., & Nguyen, V. T. (2009). Optimal of some culture conditions for Aspergillus niger VTCC-F-017 and Aspergillus oryzae VTCC-F-187 producing xylanase. In The analytica Vietnam conference 2009 (pp. 294–301).
Silva, P. O., Alencar-Guimaraes, N. C., Serpa, J. D. M., Masui, D. C., Marchetti, C. R., Verbisck, N. V., Zanoelo, F. F., Ruller, R., & Giannesi, G. (2019). Application of an endo-xylanase from Aspergillus japonicus in the fruit juice clarification and fruit peel waste hydrolysis. Biocatalysis and Agriculture Biotechnology, 21, 101312.
Miao, Y., Li, J., Xiao, Z., Shen, Q., & Zhang, R. 2015. Characterization and identification of the xylanolytic enzymes from Aspergillus fumigatus Z5. BMC Microbiology, 15.
Amir, A., Arif, M., & Pande, V. (2013). Purification and characterization of xylanase from Aspergillus fumigatus isolated from soil. African Journal of Biotechnology, 12(20), 3049–3057.
Nair, S. G., Sindhu, R., & Shashidhar, S. (2008). Purification and biochemical characterization of two xylanases from Aspergillus sydowii SBS 45. Applied Biochemistry Biotechnology, 149, 229–243.
Do, T. T., & Quyen, D. T. (2010). Purification and properties of a thermoactive xylanase from Aspergillus oryzae DSM1863. Middle East Journal of Scientific Research, 6(4), 392–397.
Krisana, A., Rutchadaporn, S., Jarupan, G., Lily, E., Sutipa, T., & Kanyawim, K. (2005). Endo-1,4-β-xylanase B from Aspergillus cf. niger BCC14405 isolated in Thailand: Purification, characterization and gene isolation. Journal of Biochemistry and Molecular Biology, 38, 17–23.
Camacho, N. A., & Aguilar, O. G. (2003). Production, purification, and characterization of a low-molecular-mass xylanase from Aspergillus sp. and its application in baking. Applied Microbiology and Biotechnology, 104, 159–172.
Fialho, M., & Carmona, E. C. (2004). Purification and characterization of xylanases from Aspergillus giganteus. Folia Microbiological (Praha), 49, 13–18.
Lu, F., Lu, M., & Lu, Z. (2008). Purification and characterization of xylanase from Aspergillus ficuum AF-98. Bioresource Technology, 99, 5938–5941.
Fernandez-Espinar, M., Pinaga, F., Graaff, L., Visser, J., Ramon, D., & Vallds, S. (1994). Purification, characterization and regulation of the synthesis of an Aspergillus nidulans acidic xylanase. Applied Microbiology and Biotechnology, 42, 555–562.
Paës, G., Berrin, J. G., & Beaugrand, J. (2012). GH11 xylanases: Structure/function/properties relationships and applications. Biotechnology Advances, 30(3), 564–592.
Ho, H. L. (2017). Aspergillus xylanases. Journal of Advances in Microbiology, 5, 1–12.
Han, N., Miao, H., Ding, J., Li, J., Mu, Y., Zhou, J., & Huang, Z. (2017). Improving the thermostability of a fungal GH11 xylanase via site-directed mutagenesis guided by sequence and structural analysis. Biotechnology for Biofuels, 10, 133.
Saleem, A., Waris, S., Ahmed, T., & Tabassum, R. (2020). Biochemical characterization and molecular docking of cloned xylanase gene from Bacillus subtilis RTS expressed in E. coli. International Journal of Biological Macromolecules, 168(2021), 310–321.
Heinen, P. R., Bauermeister, A., Ribeiro, L. F., Messias, J. M., Almeida, P. Z., Moraes, L. A. B., Vargas-Rechia, C. G., Oliveira, A. H. C., Ward, R. J., Filho, E. X. F., Kadowaki, M. K., Jorge, J. A., & Polizeli, M. L. T. M. (2018). GH11 xylanase from Aspergillus tamarii Kita: Purification by one-step chromatography and xylooligosaccharides hydrolysis monitored in real-time by mass spectrometry. International Journal of Biological Macromolecules, 108, 291–299.
Chen, Z., Zaky, A., Liu, Y., Chen, Y., Liu, L., Li, S., & Jia, Y. (2019). Purification and characterization of a new xylanase with excellent stability from Aspergillus flavus and its application in hydrolyzing pretreated corncobs. Protein Expression and Purification, 154, 91–97.
Mamo, G., Thunnissen, M., Hatti-Kaul, R., & Mattiasson, B. (2009). An alkaline active xylanase: Insights into mechanisms of high pH catalytic adaptation. Biochimie, 91, 1187–1196.
Boucherba, N., Gagaoua, M., Bouanane-Darenfed, A., Bouiche, C., Bouacem, K., Kerbous, M. Y., Maafa, Y., & Benallaoua, S. (2017). Biochemical properties of a new thermo-and solvent-stable xylanase recovered using three phase partitioning from the extract of Bacillus oceanisediminis strain SJ3. Bioresources and Bioprocessing, 4, 29.
Do, T. T., Quyen, D. T., & Dam, T. H. (2012). Purification and characterization of an acid- stable and organic solvent-tolerant xylanase from Aspergillus awamori VTCC-F312. ScienceAsia, 38(2012), 157–165.
Gaur, R., Tiwari, S., Rai, P., & Srivastava, V. (2015). Isolation, production, and characterization of thermotolerant xylanase from solvent tolerant Bacillus vallismortis RSPP-15. International Journal of Polymer Science, 2015, 1–10.
Sanghvi, G., Jivrajani, M., Patel, N., Jivrajani, H., Bhaskara, G. B., & Patel, S. (2015). Purification and characterization of haloalkaline, organic solvent stable xylanase from newly isolated halophilic bacterium-OKH. International Scholarly Research Notices.
Shu, N., Zhou, T., & Hovmöller, S. (2008). Prediction of zinc-binding sites in proteins from sequence. Bioinformatics, 24, 775–782.
Acknowledgements
This study was supported by the National Foundation for Science and Technology Development Vietnam (Nafosted), project 106.02−2018.347” Engineering of recombinant Aspergillus niger to produce highly active xylanase for functional foods industry” 2019−2021.
Author information
Authors and Affiliations
Contributions
DTT and DTMA designed the experimental setup and manuscript preparation. NTC and DTMA performed experiments of purification of xylanase. NTT and NPDN performed and characterized xylanase. NTT and DTT performed and evaluated data analysis of LC/MS analysis of purified xylanase. DTT initiated the project, read and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dao, T.M.A., Cuong, N.T., Nguyen, T.T. et al. Purification, Identification, and Characterization of a Glycoside Hydrolase Family 11-Xylanase with High Activity from Aspergillus niger VTCC 017. Mol Biotechnol 64, 187–198 (2022). https://doi.org/10.1007/s12033-021-00395-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-021-00395-8