
Abstract
Aspergillus niger van Tieghem LPM 93 was shown in an earlier study to produce the most thermostable β-xylanase, which was effective for improving brightness and delignification of non-delignified and oxygen-bleached samples of eucalyptus kraft pulp. Here, we report the production, purification, and characterization of a xylan-degrading enzyme (XynI) from this strain grown in submerged liquid cultivation on medium containing sugar cane bagasse as the carbon source. XynI was isolated by ultrafiltration and gel-filtration chromatography and characterized. The fungus displayed high levels of xylanolytic activity after the second day of cultivation, and this activity remained constant up to the 50th day. The molecular mass of XynI was in the range of 32–33 kDa as determined by mass spectrometry and SDS-PAGE. The two-dimensional gel electrophoresis analysis showed the existence of multiple forms of β-xylanases in XynI. XynI showed the highest activity at 50°C and pH 4.5 and was stable in sodium acetate buffer at pH 4.5. The K m and V max values were 47.08 mg/ml and 3.02 IU/ml, respectively. Salts inhibited the activity of XynI to different degrees. N-Bromosuccinimide caused marked inhibition of XynI. On the other hand, β-mercaptoethanol and l-tryptophan were the best enzyme activators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass is an important source of renewable energy. It consists primarily of the carbohydrate polymers cellulose and hemicellulose and the phenolic polymer lignin [21, 44]. Hemicellulose refers to a large group of heterogeneous polysaccharides. These polysaccharides possess a great variety of substituents, including sugars, in their side chains [44]. According to its structural complexity, hemicellulose hydrolysis requires an enzymatic pool composed of endo-1,4-β-d-xylanases (EC 3.2.1.8), 1,4-β-d-xylosidases (EC 3.2.1.37), α-l-arabinofuranosidases (EC 3.2.1.55), α-d-glucuronidases (EC 3.2.1.139), and acetyl-xylan esterases (EC 3.1.1.72) [5, 16, 32, 39].
Xylans are a major component of agroindustrial byproducts and waste that represent rich carbon sources for the growth of filamentous fungi and for the production of lignocellulolytic enzymes [4, 47]. Sugar cane (Saccharum officinarum) is an important commodity for many developing countries such as Brazil and India, the two biggest producers of sugar cane in the world [17]. In this context, sugar cane bagasse (SCB) is the largest Brazilian agroindustrial waste, amounting to approximately 217–380 × 109 kg/year. Although part of the bagasse is employed for internal energy generation in the sugar cane mills, some 20% of it is not used [17, 19]. The bagasse piles have low economic value and represent an environmental problem due to the risk of spontaneous combustion. A carbon source is an essential component for fermentation by microorganisms, influencing their metabolism and cellular growth [23]. SCB is an economically viable alternative carbon source for the production of industrial enzymes from filamentous fungi, bacteria, and yeasts. The enzyme described in this study provides a potential to reduce the amount of agroindustrial waste that is generated in many countries as well as to develop essential green technologies.
β-Xylanases are glycosyl hydrolases (GH) known to hydrolyze the polysaccharides from lignocellulosic biomass [5, 31]. Most of the fungal β-xylanases belong to the GH10 and GH11 families. The enzymes belonging to the GH10 family show some catalytic versatility and have higher molecular masses and lower isoelectric points than those from the GH11 family, which can efficiently hydrolyze highly branched xylans and have lower molecular weights and higher pI values [18].
Many microorganisms are capable of producing β-xylanases [32, 43, 44]. Among these, filamentous fungi are particularly promising for industry because they secrete large amounts of β-xylanases into the environment, eliminating the need for cell lysis [12, 21]. Many industrial processes can be developed using fungi or other microorganisms as enzyme sources and, in many cases, the efficiency can be improved by using pure enzymes [4]. The fungus Aspergillus niger is widely used in many biotechnological processes including biopulping, biorefineries, food and pharmaceutical industries [30]. The most important advantages associated with its use are its safety for humans during enzyme production [35] and its versatile metabolism, allowing its growth on many substrates and under many environmental conditions [33].
In two previous publications [26, 27], ten fungal species were isolated from decomposed wood in the natural forest reserve of National Research Institute of Amazonia (Brazil), purified, and evaluated for their capacity to produce xylan-degrading enzyme activity during growth in liquid medium containing oat-spelt xylan as the carbon source. A. niger van Tieghem LPM 93 was the most efficient at producing thermostable β-xylanase [27]. The crude xylanase preparation from A. niger van Tieghem LPM 93 was effective for improving brightness and delignification of non-delignified and oxygen-bleached samples of eucalyptus kraft pulp [27]. The aim of the present study was to isolate and characterize a xylan-degrading enzyme (XynI) produced by the mesophilic fungus A. niger van Tieghem LPM 93 when grown by submerged liquid cultivation (SLC) containing SCB as carbon source.
Materials and Methods
Chemicals
All substrates, N-bromosuccinimide (NBS), dithiothreitol (DTT), 5,5-dithio-bis(2-nitrobenzoic acid) (DTNB), 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDC), diethyl pyrocarbonate (DEPC) and 2,2′-dithiopyridine (DTP), oat-spelt xylan, carboxymethyl cellulose (CMC), polygalacturonic acid, galactomannan, microcrystalline cellulose (avicel), p-nitrophenyl-β-d-xylopyranoside (pNPX), p-nitrophenyl-β-d-glucopyranoside (pNPG) and p-nitrophenyl-α-l-arabinofuranoside (pNPA) were purchased from Sigma Aldrich Chemical Co. Chromatography resins and filter paper (Whatman no 1) were from GE Healthcare. SCB (S. officinarum L., variety Java) was from a local source.
Residue Pretreatment
SCB (S. officinarum L., variety Java) was ground in a bench grinder, thoroughly washed with tap water and autoclaved at 121°C for 2 h. After being autoclaved, it was dried at 65°C for 48 h and ground to form a homogeneous blend. A fine powder was obtained and used as a substrate for the fungus.
Enzyme Production
A. niger van Tieghem LPM 93 was obtained from the fungus culture collection of the Enzymology Laboratory, University of Brasília, Brazil and was maintained in PDA medium (2.0% potato broth, 2.0% dextrose, and 2.0% agar) at 28°C and cultured on SCB. The basal culture medium composition (g/l) was as follows: 7.0 g KH2PO4, 2.0 g K2HPO4, 0.1 g MgSO4.7H2O, 1.0 g (NH4)2SO4, 0.6 g yeast extract and 1% of SCB at pH 7.0. A portion (5.0 ml) of an A. niger van Tieghem LPM 93 spore suspension (108 spores/ml) was introduced into an Erlenmeyer flask (2 l) containing 500 ml of liquid medium with agroindustrial residue as the carbon source. SLC was carried out at a substrate concentration of 1.0% (w/v) for 6 days at 28°C with agitation at 120 rpm. After the culture had grown, the medium was passed through filter paper (Whatman No. 1). The resulting filtrate, hereafter called crude extract, was stored at 5°C and used for further isolation and characterization of the β-xylanase samples. For β-xylanase induction, aliquots were harvested every 24 h during 50 days and used to estimate the enzyme activity and protein concentration.
Enzyme Purification
The crude extract was concentrated approximately 10-fold by ultrafiltration using an Amicon System (Amicon Inc., Beverly, MA 01915, USA) with a membrane having a cutoff point of 10 kDa (PM 10) at 10°C and 2.5 kgf/cm2. Aliquots (500 ml) of the ultrafiltrate were precipitated with 60% (w/v) saturation of ammonium sulfate and allowed to settle for 15 h at 5°C. The precipitate was obtained by centrifugation at 4,500×g for 20 min at 4°C and dissolved in 50 ml of 50 mM sodium phosphate buffer, pH 7.0. It was designated as UFPM10. Aliquots of UFPM10 (18 ml) were fractionated by gel-filtration chromatography on a Sephadex G-50 (2.7 × 60.0 cm) column pre-equilibrated with 50-mM sodium phosphate buffer, pH 7.0, containing 0.15 M NaCl. Fractions (5 ml) were collected at flow rate of 20 ml/h, and those corresponding to β-xylanase activity, hereafter named XynI, were pooled and stored at 5°C.
Enzymatic Assays
Endoglucanase, β-xylanase, polygalacturonase and mannanase activities were determined by mixing 50 μl of enzyme sample with 100 μl of 1% (w/v) substrate (CMC, oat-spelt xylan or polygalacturonic acid, sodium salt) or 0.5% (w/v) substrate (galactomannan) at 50°C for 30 min, respectively. Filter paper activity (FPase) [25] was determined using 150 μl of enzyme with filter paper as the substrate at 50°C for 1 h. Avicelase activity was determined by mixing 50 μl of avicel suspension (1%, w/v) with 100 μl of enzyme sample at 50°C for 2 h. The amount of reducing sugar released was measured using dinitrosalicylic reagent [28]. Activity was expressed as micromoles of reducing sugar formed per minute per milliliter of enzyme solution, i.e., as IU/ml. Glucose, xylose, mannose, and galacturonic acid were used as standards. β-Xylosidase, β-glucosidase, and α-arabinofuranosidase activities were determined with the substrates pNPX, pNPG, and pNPA, respectively [45, 46]. Protein concentration was determined by the Bradford assay [3] using bovine serum albumin as a standard.
Enzyme Characterization
The influence of the temperature on β-xylanase activity was measured by performing the standard activity assay at temperatures ranging from 30°C to 70°C. The temperature stability of β-xylanase was determined by pre-incubating the enzyme samples at 45°C, 50°C, and 55°C and removing samples at intervals to measure the activity as described before. The enzyme stability was also measured using 50 mM sodium acetate buffer, pH 4.5 at 45°C and 50°C, and in the presence of l-tryptophan or β-mercaptoethanol at a final concentration of 10 mM at 45°C. The influence of pH on β-xylanase activity was assessed by incubating 25 μl of enzyme solution, 50 μl of xylan (1%, w/v), and 75 μl of each the following buffers: 50 mM sodium acetate (pH 3.0–6.0), 50 mM sodium phosphate (pH 6.0–7.5), or 50 mM Tris–HCl (pH 7.5–9.0), respectively, at 45°C and 50°C. All buffers, regardless of pH, were adjusted to the same ionic strength with NaCl. The effects of several salts (MgCl2, MgSO4.7H2O, AlCl3, HgCl2, NaCl, ZnSO4, CaCl2, KCl, FeCl3, FeSO4, CuSO4, MnCl2, CuCl2, AgNO3, and CoCl2) and other agents (DTP, DTNB, EDC, DEPC, l-tryptophan, l-cysteine, iodoacetamide, DTT, β-mercaptoethanol, NBS, SDS, and EDTA) on β-xylanase activity were tested after 30 min of incubation at 29°C in the presence of the individual reagents at final concentrations in the range of 0.5–10 mM, followed by the standard β-xylanase assay under the following conditions: 25 μl of XynI, 75 μl of the reagent, and 50 μl of xylan. For the kinetic experiments, soluble and insoluble xylans from oat spelt were used as substrates in concentration ranges of 4–50 and 0.5–6.0 mg/ml, respectively. The substrates were saturating and the enzyme activities were proportional to the amount of enzyme added. Soluble and insoluble xylans were prepared as described by Filho et al. [8, 9]. K m and V max values were estimated from the Michaelis–Menten equation with a nonlinear regression data analysis program [24]. Each assay described above was repeated at least three times; the standard deviation was less than 20% of the mean.
Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of β-xylanase samples were carried out as described by Laemmli [22] using 12% gels. After electrophoresis, the protein bands were silver stained by the method of Blum et al. [2]. For the detection of β-xylanase activity, zymograms were carried out as described by Wang et al. [43]. Replicate denaturing electrophoretic gel, containing 0.1% oat-spelt xylan, was submitted to zymogram analysis. It was stained for β-xylanase activity in a Congo red solution (0.5 mg/ml) for 15 min at room temperature and washed with 1 M NaCl to remove excess dye and fixed with 1 M HCl. The molecular mass of XynI was estimated by SDS-PAGE using low molecular mass markers (GE Healthcare). For two-dimensional gel electrophoresis the samples were previously treated with the 2D-Clean-Up Kit (GE Healthcare) and resuspended in 350 μl of solution containing DTT (85 mM), Triton X-100 (2.5%, w/v), IPG buffer at pH 3–10 (GE® 0.5%, w/v), urea (7 M), thiourea (2 M), and isopropanol (10%). The samples were applied to 18 cm pH 3–10 linear immobilized pH gradient strips (Immobiline™ Dry Strips, GE Healthcare) by in gel rehydration and analyzed by isoelectric focusing on an Ettan IPGphor III apparatus (GE Healthcare). The second dimension (8–15% polyacrylamide gradient, SDS-PAGE) was carried out in BioRad Protean® II xi Cells.
Mass Spectrometry
Protein spots detected on the two-dimensional gel electrophoresis of the XynI purified fraction were excised, reduced with DTT, alkylated with acrylamide, and digested with trypsin (Promega, Madison, USA) as previously described [29]. Protein digests were analyzed by peptide mass fingerprinting (PMF) and peptide fragment fingerprinting by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry using an Autoflex II MALDI-TOF-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). For analysis, 2 μl of each digest was mixed with 1 μl of matrix (10 μg/μl α-cyano-4-hydroxycinnamic acid in 70% (v/v) acetronitrile, 0.1% (w/v) TFA) on the surface of an AnchorChip™ plate (Bruker). External calibration was performed using a peptide standard kit (Bruker Daltonics). Known trypsin autolysis and keratin peaks were used for internal calibration. Peptide masses (MH+) were recorded in 750 to 3,000 Da range. The peptide mass spectra were generated using the software FlexControl v. 2.4 (Bruker Daltonics). The same software was used to acquire and process the peak lists that was employed for database searches using BioTools v. 2.0 (Bruker Daltonics) linked to Mascot (http://www.matrixscience.com/) against the National Center for Biotechnology Information protein database (NCBI; Bethesda, USA). Monoisotopic masses of tryptic peptides were used to identify the proteins by PMF. Error tolerance for peptide mass was lower than 100 ppm and no restrictions were imposed on protein molecular mass. Further search parameters were: one missed cleavage site for trypsin, methionine oxidation as a variable modification and propionamide cysteine (acrylamide alkylation) as a fixed modification. Hits were considered significant if the protein score exceeded the threshold score calculated by Mascot software assuming a p value of <0.05.
Results and Discussion
Induction Profile
The induction profile during growth of A. niger van Tieghem LPM 93 on SCB showed that β-xylanase activity increased steadily without a lag and reached a plateau that lasted from the second day to the end of the cultivation period. The growth profile was accompanied by several peaks of protein. A multiplicity of forms is commonly described for β-xylanases from fungi and bacteria as result of differential mRNA processing and posttranslational modifications [44]. This profile of induction suggests a progressive access to the hemicellulose structures that permeate the cellulose fibers of SCB, stimulating the production and gradual release of hydrolytic enzymes for the consumption of the substrate. The presence of soluble sugars in the culture medium apparently did not significantly inhibit the production of β-xylanases, but it may have been responsible for maintaining the enzyme activity without significant variation from the second day of growth of the fungus. It is also possible that the sugar released into the environment was used by the fungus as an energy source because there was no nutrient addition during the period studied. The medium collected on the sixth day of growth contained a protein peak that coincided with a peak of high xylanolytic activity. The amount of total protein varied during the growth period studied. This protein profile probably includes other proteins, in addition to β-xylanases, which are simultaneously produced and may be involved in the complex process of SCB degradation. Therefore, based on the growth curve of the fungus and in order to obtain large amounts and a high diversity of xylanolytic enzymes, we established six days for fungal growth in liquid medium containing SCB.
The influence of SCB on the synthesis of β-xylanase was examined by electrophoresis under denaturing conditions (data not shown). The SDS-PAGE of the crude extract samples from the inducing medium revealed protein bands with molecular weights ranging from 14 to 90 kDa. A pronounced protein band of approximately 30 kDa was detected between 2 and 16 days of incubation. It was coincident with bands that stained for β-xylanase activity after zymogram analysis (Fig. 1c). After the 1st day of incubation, a protein band with high molecular weight (above 66 kDa) could be seen. Protein bands with low molecular weight (less than 14 kDa) were only detected after the 6th day of incubation.

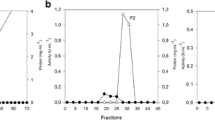
a Chromatographic profile of UFPM10 in a Sephadex G-50 column. Total protein (solid line) and xylanolitic activity (dashed line). b SDS-PAGE (12%) of the purification steps of the crude extract from A. niger van. Tieghem LPM 93 grown on liquid medium containing SCB. Line 1, markers; line 2, crude extract; line 3, UFPM10; line 4, XynI. c Zymogram: line 1, crude extract; line 2, UFPM10; line 3, XynI
Enzyme Purification
The pool of xylanolytic enzymes obtained from the SLC containing SCB as the carbon source was isolated by a combination of ultrafiltration, ammonium sulfate precipitation and chromatographic procedures. The crude extract was concentrated 10-fold by ultrafiltration. β-Xylanase activity was found in the retentate and ultrafiltrate. The amount of protein of ultrafiltrate (0.5 mg) was much lower than the retentate (25.5 mg). The xylanase activity of the ultrafiltrate and concentrate were 0.5 and 1.20 IU/ml. For further purification, the ultrafiltrate was precipitated with 60% of ammonium sulfate saturation. The β-xylanase activity was only found in the precipitate, which, in turn, was fractionated by gel-filtration chromatography on Sephadex G-50 column (Fig. 1a). A single peak of β-xylanase activity was eluted before a major peak of protein. The purification procedure provided a yield of 9.5% and a 14.9-fold purification. Since other forms of β-xylanase were detected in the retentate, and these enzymes may act synergistically to effect xylan breakdown, the fold purification, and recovery values were underestimated [46]. This phenomenon is often described during purification of β-xylanases produced by fungi. Teixeira et al. [42] reported yield and fold purification of 4.58 and 16.88, respectively for β-xylanase of Aspergillus awamori. The ultrafiltration procedure retained most of the the β-xylanase activity in the retentate. Moreover, comparison of these values with those reported for the β-xylanases from other sources is not very meaningful because of the high interlaboratory variability in assays, and because β-xylanases differ from one another with respect to their mechanism of action. The apparent purity of the enzyme was demonstrated by SDS-PAGE and zymogram analysis (Figs. 1b, c). The gel under denaturing conditions showed a single band. The molecular mass of XynI was found to be in the range of 32–33 kDa, as estimated by SDS-PAGE. This is in agreement with the range determined more accurately for the native enzyme by using mass spectrometry, a value range that compares well to previously reported data on A. awamori xylanase [42]. A single peak was detected on the mass spectrum (Fig. 2). Those results revealed the ability of β-xylanases to change their conformation and pass through membranes with a cutoff of 10 kDa [9, 36, 40]. The ability to pass through the small pores in the wood and thus to penetrate the hemicellulose-lignin-cellulose matrix could be advantageous, especially for filamentous fungi [11], and this property could be explored for biotechnological applications. In support of the SDS-PAGE result, zymogram analysis revealed one β-xylanase activity band coincident with that staining for protein. A clear hydrolysis activity zone was formed against a dark background (Fig. 1c).

MALDI-TOF spectrum of XynI
Enzyme Characterization
The substrate specificity of XynI was restricted to xylan. It was devoid of measurable pectinase, mannanase, cellulase, β-xylosidase, α-arabinofuranosidase and β-glucosidase activities. The specificity of XynI for xylan as substrate is an important parameter for its use in pulp bleaching, whereas in this process the enzyme has to be cellulase free. The rate dependence of the β-xylanase reaction on soluble and insoluble xylans followed Michaelis–Menten kinetics. Nonlinear regression data analysis determination of kinetic parameters of XynI acting on soluble and insoluble oat-spelt xylans showed that the enzyme had affinity only for soluble xylans, with K m and V max values of 47.08 mg/ml and 3.02 IU/ml, respectively, suggesting that the presence of a particular type of substituent (arabinofuranosyl group) in the vicinity would be a requirement for the action of XynI. In this case, the substituent (arabinofuranosyl residue) may be required for the proper orientation of xylan in the catalytic site. Consistent with this possibility is the fact that XynI was not active against insoluble xylan. Thus, the absence of such branches in the insoluble xylan could prevent the adsorption of XynI to the substrate. Conversely, the hydrolysis of insoluble oat-spelt xylan by β-xylanase II from Aspergillus fumigatus was more effective than when the enzyme was incubated with soluble xylan [37]. This might suggest a steric hindrance due to the presence of substituents in soluble xylan. In comparison with the K m values of some β-xylanases [8, 9, 37], Xyl showed lower affinity for soluble xylan. Nevertheless, enormous variations in kinetic parameter values have been reported for β-xylanases from various microorganisms. These variations may be attributed to differences in assay procedures [14, 34, 41]. The type of substrate has a significant effect on these values.
Generally speaking, β-xylanases from fungal sources are reported to be more active and stable in the temperature range of 40–55°C under acidic conditions [41]. In addition, a comparison of temperature effects on β-xylanases from Aspergillus spp. [39] showed that for most naturally occurring β-xylanases the activity was highest in the temperature range of 45–60°C. Other studies show that the best temperature for β-xylanase activity depends on the type of carbon source used for growing the fungus. Medeiros et al. [26] demonstrated that the β-xylanase from a crude extract of A. niger van Tieghem LPM 93 previously cultivated in liquid media containing xylan reached its highest value at 40°C. β-Xylanase activity isolated from a crude extract of the same fungus grown on wheat bran was most active at 48°C [48]. Solid state cultivation of A. niger in sugar cane bagasse showed a β-xylanase with maximum activity at 35°C [10]. In the present study, the crude extract, UFPM10 and XynI samples were most active between 45°C and 50°C. Within this temperature range, XynI and the crude extract displayed a higher yield of β-xylanase activity at 50°C. However, at 45°C UFPM10 showed the best yield of β-xylanase activity. The pH profile of crude extract, UFPM10 and XynI samples showed that β-xylanase activity remained significant in acidic conditions. It displayed high activity over a broad pH range (3.5–5.5), being most active at pH 4.5. β-Xylanases of many species of the genus Aspergillus are most active in the pH range of 4.0–6.0 [39, 41]. As described in this paper, there are some other exceptions such as β-xylanases from A. kawachii and A. niger, which exhibit higher activity at pH 2.0–6.0 and 3.0, respectively [15, 20]. The effect of sodium acetate buffer on the thermostability of XynI, crude extract and UFPM10 was determined at 45°C and 50°C. For the purpose of comparison, we used aqueous solutions of XynI, crude extract and UFPM10. XynI and crude extract were stable at 45°C and pH 4.5 with half-lives of 110 and 144 h, respectively. UFPM10 was less stable with a half-life of 36 h. Compared with the crude extract and UFPM10, XynI was less stable at 45°C and 50°C in the absence of sodium acetate buffer, with half-lives of 48 h and 10 min, respectively. The effects of temperature and pH on enzymatic activity are important parameters for determining the type of industrial application of enzyme. The acid tolerant property of XynI, crude extract and UFPM10 samples show the potential for their use in industrial processing, especially in the fruit and textile industries. For XynI, we may predict that it does not possess the commonly necessary characteristics for applications in the pulp and paper industry, like tolerance to alkaline pH and high temperatures [27]. However, XynI and the other samples may be used in pulp bleaching processes that require moderate temperature and acid pH [1]. It has been more frequently observed the use of β-xylanases with highest activity in pH below 5.5 [38]. Some commercial β-xylanases that are used in the textile industry, especially for treating cotton fibers, exhibit their highest activity in the pH range of 4.5–5.0 and at 50°C [6]. Another possibility for the use of β-xylanases is in the brewing industry due to the ability of this enzyme to replace the additives traditionally used as emulsifiers and oxidants.
The effects of several reagents on XynI activity are summarized in Table 1. Most of the metal ions, including Al3+ and Cu2+, inhibited the activity of XynI to different degrees. The enzyme was strongly inhibited by Hg2+ at 10 mM concentration. The inhibition of XynI by a sulfhydryl oxidant metal (Hg2+) may be due to complex formation with and (or) catalysis of oxidation of specific residues (thiol groups), or nonspecific salt formation. SDS was also a potent inhibitor of XynI at concentrations of 2 and 10 mM. The involvement of some amino acid modifying agents on XynI activity was investigated (Table 2). XynI was highly activated by l-tryptophan and β-mercaptoethanol with increases of 88.82% and 82.70% of its activity, respectively. The treatment of XynI with DTT, DTNB and iodoacetamide activated the enzyme activity, suggesting an influence of l-cysteine in the catalysis of xylan. l-Cysteine is thought to be involved in hydrogen-bonding with the substrate and may be involved with enzyme folding and the formation of the covalent glycosyl-xylan intermediate [7]. XynI activation by DTT suggests that reduction of disulfide bridge(s) probably oxidized during enzyme extraction, and that purification procedures restored the native XynI conformation. Contradicting what was previously described [42], l-cysteine inhibited the activity of XynI. This suggests that l-cysteine may not be specific, which means that some essential but inaccessible groups may not be modified by the agent used, and modification of groups at a distance from the active site may affect conformational changes and consequently cause loss of β-xylanase activity [7]. XynI was strongly inhibited (75%) by NBS, a potent oxidizing agent of l-tryptophan. The indole ring of tryptophan is a reactive functional group in proteins and is modified by many electrophilic and oxidizingt reagents [13]. This effect has been previously reported for β-xylanase activities from different fungus species, suggesting the involvement of l-tryptophan in substrate catalysis [8, 9].
The involvement of l-tryptophan and β-mercaptoethanol on the temperature stability of β-xylanase activity from the crude extract, UFPM10 and XynI samples was investigated. L-Tryptophan and β-mercaptoethanol were not able to protect the high β-xylanase activity in samples of the crude extract for a long time. For the same sample, incubation with sodium acetate buffer, pH 4.5, increased the half-life of β-xylanase 14.4 and 24 times when compared to incubation with l-tryptophan and β-mercaptoethanol, respectively. The buffer caused the same effect in XynI, increasing its half-life 27.5 times. On the other hand, the incubation of UFPM10 with l-tryptophan was more effective than with sodium acetate buffer and β-mercaptoethanol, increasing the β-xylanase half-life in 1.5 times. This indicates that the purification process of XynI may have removed cofactors present in UFPM10 but absent in XynI. In the crude extract sample, which contains other enzymes besides β-xylanase, the influence of these cofactors was probably less.
The two-dimensional profile of proteins secreted by A. niger van Tieghem LPM 93 in the presence of SCB shows that most of the proteins are found in the acidic range of the gel, and that there is a predominance of high molecular mass proteins that were removed from the crude extract during the purification process of XynI (Figs. 3a, b). The presence of spots with slight differences of molecular mass and pI values shows the existance of isoforms or multiple forms in XynI sample. Peptide mass fingerprinting and peptide fragment fingerprinting analysis of the spots 1, 3, and 4, present in the XynI two-dimensional profile, indicate that the first one matched an α-arabinofuranosidase from A. tubingensis (NCBI access number gi 3913152). It is important to remember that XynI only had affinity for soluble xylan. This might suggests that XynI could have a bifunctional catalytic role, liberating free arabinose in addition to cleaving the main chain linkages of arabinoxylan. The two other spots matched endo-1,4-β-xylanases from A. aculeatus (NCBI access number gi 3915310). The other spots present in XynI two-dimensional gels could not be identified.

Two-dimensional electrophoresis of proteins secreted by A. niger van Tieghem LPM 93 in the presence of SCB. a Crude extract and b XynI. The arrows 1–4 indicate the spots that were selected for digestion but only the spots 1 (A. tubingensis α-l-arabinofuranosidase), 3 and 4 (A. aculeatus endo-1,4-β-xylanases) were identified
Conclusions
Finding enzymes which operate at desirable pH and temperature for a specific industrial application is a challenging task. In this study, XynI showed acid tolerance and stability at 45°C. Analysis of XynI by two-dimensional electrophoresis indicates the presence of isoforms. Further studies will also focus on the isolation and characterization of those isoforms, including the determination of mechanism of action, the use of atomic force microscopy as a tool to study the three-dimensional structure and possible topographical differences among the isoforms and glycosylation degree.
References
Bissoon S, Singh S, Christov L (2002) Evaluation of the bleach-enhancing effect of xylanases on bagasse pulp. Progress Biotechnol 2:247–254
Blum H, Beier H, Gross B (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrilamide gels. Electrophoresis 8:93–99
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chidi SB, Godana B, Ncube I, van Rensburg EJ, Cronshaw A, Abotsi EK (2008) Production, purification and characterization of cellulase-free xylanase from Aspergillus terreus UL 4209. African J Biotechnol 7:3939–3948
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xilanases. FEMS Microbiol Rev 29:3–23
Csiszár E, Urbánszki K, Szakács G (2001) Biotreatment of desized cotton fabric by commercial cellulase and xylanase enzymes. J Mol Catalysis B: Enzymatic 11:1065–1072
Ferreira HM, Filho EXF (2004) Purification and characterization of a β-mannanase from Trichoderma harzianum strain T4. Carbohydr Polym 57:23–29
Filho EXF, Puls J, Coughlan MP (1993) Biochemical characteristics of two endo-β-1,4-xylanases produced by Penicillium capsulatum. J Ind Microbiol Biotechnol 11:171–180
Filho EXF, Puls J, Coughlan MP (1993) Physicochemical and catalytic properties of a low-molecular-weight endo-1,4-β-d-xylanase from Myrothecium verrucaria. Enzyme Microb Technol 15:535–540
Gawande PV, Kamat MY (1999) Production of Aspergillus xilanases by lignocellulosic waste fermentation and its application. J Appl Microbiol 87:511–519
Grabski AC, Jeffries TW (1991) Production, purification, and characterization of β-(1,4)-endoxylanase of Streptomyces roseiscleroticus. Appl Environ Microbiol 57:987–992
Haltrich D, Nidetzky B, Kulbe KD, Steiner W, Zupancic S (1996) Production of fungal xylanases. Biores Technol 58:137–161
Imoto T, Yamada H (1990) Chemical modification. In: Creighton TE (ed) Protein function, a practical approach. IRL Press, Oxford, pp 247–278
Ishihara M, Tawata S, Toyama S (1997) Purification and some properties of a thermostable xylanase from thermophilic fungus strain HG-1. J Ferment Bioeng 83:478–480
Ito K, Ogassawara J, Sugimoto T, Ishikawa T (1992) Purification and properties of acid stable xylanases form Aspergillus kawachii. Biosc Biotechnol Biochem 56:547–550
Jovanovic I, Magnuson JK, Collart F, Robbertse B, Adney WS, Himmel ME et al (2009) Fungal glycoside hydrolases for saccharification of lignocellulose: outlook for new discoveries fueled by genomics and functional studies. Cellulose 16:687–697
Kadam KL (2002) Environmental benefits on a life cycle basis of using bagasse-derived ethanol as a gasoline oxygenate in India. Energy Policy 30:371–384
Kapoor M, Singh A, Kuhad RC (2007) Application of xylanases in the pulp and paper industry: an appraisal. In: Kuhad RC, Singh A (eds) Lignocellulose biotechnology: future prospects. IK International, New Delhi, pp 307–310
Khan MA, Ashraf SM, Malhotra VP (2004) Development and characterization of a wood adhesive using bagasse lignin. Int J Adhesion and Adhesives 24:485–493
Krengel U, Dijkstra BW (1996) Three-dimensional structure of endo-1,4-β-xylanase I from Aspergillus niger: molecular basis for its low pH optimum. J Mol Biol 263:70–78
Kulkarni N, Shendye A, Rao M (1999) Molecular and biotechnological aspects of xilanases. FEMS Microbiol Rev 23:411–456
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Laxmi GS, Sathish T, Rao CS, Brahmaiah P, Hymavathi M, Prakasham RS (2008) Palm fiber as novel substrate for enhanced xylanase production by isolated Aspergillus sp. RSP-6. Curr Trends Biotechnol Pharm 2:447–455
Leatherbarrow RJ (1999) Enzfitter Manual, a non-linear curve fitting program for Windows. Biosoft, London, pp 1–104
Mandels M, Andreotii R, Roche C (1976) Measurement of saccharifying cellulase. Biotechnol Bioeng Symposium 16:21–33
Medeiros RG, Hanada R, Filho EXF (2003) Production of xylan-degrading enzymes from Amazon Forest fungal species. Int Biodet Biodegr 52:97–100
Medeiros RG, da Silva Jr FG, Báo SN, Hanada R, Filho EXF (2007) Application of xylanases from Amazon forest fungal species in bleaching of eucalyptus kraft pulps. Braz Arch Biol Technol 50:231–238
Miller G (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Paba J, Santana JM, Teixeira ARL, Fontes W, Sousa MV, Ricart CAO (2004) Proteomic analysis of the human pathogen Trypanosoma cruzi. Proteomics 4:1052–1059
Pandey A, Selvakumar P, Soccol CR, Nigam P (1999) Solid state fermentation for the production of industrial enzymes. Curr Sci 77:149–162
Parkkinen T, Hakulinen N, Tenkanen M, Siika-aho M, Rouvinen J (2004) Crystallization and preliminary X-ray analysis of a novel Trichoderma reesei xylanase IV belonging to glycoside hydrolase family 5. Acta Cryst Section D 60:542–544
Polizeli MLM, Rizzatti ACS, Monti R, Terenzi HF, Jorge JA, Amorim DS (2005) Xylanases from fungi: properties and industrial applications. Appl Microbiol Biotechnol 67:577–591
Raj HG, Saxena M, Allameh A (1992) Metabolism of foreign compounds by fungi. In: Arora DK, Elander RP, Mukerji KG (eds) Handbook of applied mycology. Marcel Dekker, New York, pp 881–904
Salama MA, Ismail KMI, Amany HA, El-Lill A, Geweely NSI (2008) Biochemical studies of purified extracellular xilanases from Aspergillus versicolor. Int J Bot 4:41–48
Schuster E, Dunn-Coleman N, Frisvad JC, van Dijck PW (2002) On the safety of Aspergillus niger—a review. Appl Microbiol Biotechnol 59:426–435
Shei JC, Fratzke AR, Frederick MM, Frederick JR, Reilly PJ (1985) Purification and characterization of endo-xylanases from Aspergillus niger. II. An enzyme of pI 4.5. Biotechnol Bioeng 27:533–538
Silva CHC, Puls J, Sousa MV, Filho EXF (1999) Purification and characterization of a low molecular weight xylanase from solid state cultures of Aspergillus fumigatus. Braz J Microbiol 30:114–119
Subramaniyan S, Prema P (2000) Cellulase-free xilanases from Bacillus and other microorganisms. FEMS Microbiol Lett 183:1–7
Subramaniyan S, Prema P (2002) Biotechnology of microbial xilanases: enzymology, molecular biology and application. Crit Rev Biotechnol 22:33–46
Tan LUL, Mayers P, Saddler JN (1987) Purification and characterization of a thermostable xylanase from thermophilic fungus Thermoascus aurantiacus. Can J Microbiol 33:689–692
Taneja K, Gupta S, Kuhad RC (2002) Properties and application of a partially purified alkaline xylanase from an alkalophilic fungus Aspergillus nidulans KK-99. Biores Technol 85:39–42
Teixeira RSS, Siqueira FG, Souza MV, Filho EXF, Bon EPS (2010) Purification and characterization studies of a thermostable β-xylanase from Aspergillus awamori. J Ind Microbiol Biotechnol 37:1041–1051
Wang P, Mason C, Broda P (1993) Xylanases from Streptomyces cyaneus: their production, purification and characterization. J Gen Microbiol 139:1987–1993
Wong KKY, Tan LUL, Saddler JN (1988) Multiplicity of β-1,4-xylanase in microorganisms: functions and applications. Microbiol Rev 52:305–317
Ximenes FA, Silveira FQP, Filho EXF (1996) Production of β-xylosidase activity by Trichoderma harzianum strains. Curr Microbiol 33:71–77
Ximenes FA, Sousa MV, Puls J, da Silva Jr FG, Filho EXF (1999) Purification and characterization of a low-molecular-weight xylanase produced by Acrophialophora nainiana. Curr Microbiol 38:18–21
Zhang YHP, Himmel ME, Mielenz JR (2006) Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv 24:452–481
Zhao J, Li X, Qu Y, Gao P (2002) Xylanase pretreatment leads to enhanced soda pulping of wheat straw. Enz Microb Tehnol 30:734–740
Acknowledgments
E.X.F.F., C.A.O.R., and L.P.S. acknowledge receipt of a research fellowship from the Brazilian Research Council (CNPq). N.G.M. and D.P.G.M. acknowledge receipt of a postgraduate maintenance scholarship from CNPq. This work was funded by the Foundation for Research Support of the Federal District (Brazil, research grant number 193.000.470/2008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
von Gal Milanezi, N., Mendoza, D.P.G., de Siqueira, F.G. et al. Isolation and Characterization of a Xylan-Degrading Enzyme from Aspergillus niger van Tieghem LPM 93 with Potential for Industrial Applications. Bioenerg. Res. 5, 363–371 (2012). https://doi.org/10.1007/s12155-011-9137-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12155-011-9137-3