
Abstract
Introduction
Lafora disease (LD) is a severe form of progressive myoclonus epilepsy characterized by generalized seizures, myoclonus, intellectual decline, ataxia, spasticity, dysarthria, visual loss, and in later stages, psychosis and dementia. To date, mutations in the EPM2A and EPM2B/NHLRC1 genes have been identified as the common causes of LD. However, a mutation in PRDM8 has been reported only once in a Pakistani family affected with early-onset Lafora disease. In the present study, we report the second family with a PRDM8 mutation.
Methods
Two affected individuals of an Iranian family initially diagnosed as complicated hereditary spastic paraplegia (HSP) underwent careful neurologic examination. Homozygosity mapping and whole-exome sequencing were performed. Based on the results of genetic analysis to detection of Lafora bodies, a skin biopsy was done.
Results
The clinical features of the patients were described. Linkage to chromosome 4 and a mutation in the PRDM8 gene were identified, suggesting the patients may be affected with early-onset LD. However, like the Pakistani family, the search for Lafora bodies in their skin biopsies was negative. Their electroencephalograms showed generalized epileptiform discharges in the absence of clinical seizures.
Conclusions
The current study increases the number of PRDM8-related cases and expands the phenotypic spectrum of mutations in the PRDM8 gene. Both reported PRDM8-related families presented intra and inter-familial heterogeneity and they have originated from the Middle East. Thus, it seems the PRDM8 mutations should be considered not only in LD but also in other neurodegenerative disorders such as a complicated HSP-like phenotype, especially in this region.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lafora disease (LD) is an autosomal recessive and severe form of progressive myoclonus epilepsy (PME) that develops at 8 to 19 years [1]. The disease is characterized by generalized seizures, myoclonus, and poor school performance in most patients. The progressive neurodegeneration and impairment of cerebral cortical neurons result in intellectual decline, ataxia, spasticity, motor dysfunction, dysarthria, visual loss, and in later stages, psychosis and dementia [2–4]. Death usually occurs within 10 years of the disease onset [3, 5, 6]. Lafora disease is a relatively rare disorder, and its prevalence is comparatively higher in the Mediterranean, Northern African, or Asian countries [7]. LD, known as a glycogen storage disease, is caused by forming an aberrant poorly branched and insoluble polymer of glucose, called polyglucosan [5, 8–10]. Therefore, it accumulates and forms Lafora bodies in different tissues such as skin and brain [11–13]. Lafora bodies play a crucial role in the pathogenesis of LD and those are not found in other types of progressive myoclonus epilepsies [14].
Until now, bi-allelic pathogenic variants in three causative genes, EPM2A (OMIM:607,566), EPM2B/NHLRC1 (OMIM:608,072), and PRDM8 (OMIM:616,640), have been associated with Lafora disease [12, 13, 15, 16]. The EPM2A and EPM2B genes encode laforin and malin, and their mutations are responsible for 58% and 35% of LD cases, respectively [10, 17]. Both genes are involved in glycogen synthesis regulation [18, 19], and their pathogenic variants result in the typical/classic forms of Lafora disease [6].
Evidence for mutation in the PRDM8 gene, located on chromosome 4q21.21, has previously been reported only once in a single consanguineous Pakistani family. The family was affected with an atypical form of LD called early-onset Lafora disease or progressive myoclonic epilepsy-10 (EPM10; OMIM:616,640) [12]. The affected individuals presented an earlier age at onset, about 5 years less than classic LD. Dysarthria, myoclonus, ataxia, and motor defects began as first symptoms in early childhood. Subsequently, they variably manifested other classic symptoms of LD [3, 12].
PRDM8 acts as a multifunctional gene [20]. It plays an important role in mice neocortical development [21] and may be a candidate gene for human congenital stationary night blindness [22]. Moreover, the protein encoded by the PRDM8 gene is a probable histone methyltransferase [12, 23, 24] and also involved in the innate immune system [25]. This protein interacts with cytoplasmic laforin and malin and relocates them to the nucleus [12]. So, it may contribute in regulation of glycogen synthesis and its abnormality results in a glycogen storage disease [12].
In the present study, we report the second family with a PRDM-related disorder. The phenotypic manifestations of our patients were not completely matched to early-onset LD patients.
Subjects and methods
This research was performed by the Declaration of Helsinki and with approval of the ethics board of the University of Social Welfare and Rehabilitation Sciences (USWR) in Iran.
Subjects
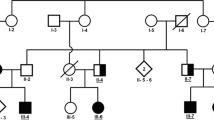
Two siblings suspected to have HSP (HSP100-IV1 and HSP100-IV2), born to the consanguineous Iranian parents, were referred to the genetics research center (GRC) of the USWR for genetic analysis (Fig. 1A). Initially, they were referred to us as “complicated HSP” cases. The mode of inheritance of disease was consistent with an autosomal (or X-linked) recessive pattern, as two male offspring were born to unaffected consanguineous parents. Clinical data from both affected siblings were collected. Relevant information on the patients not reported in Table 1 is presented in the results section.

A The Iranian Lafora disease (LD) pedigree. Genotypes of the PRDM8 variant are shown when individuals were assessed. Arrow shows proband. Unfilled circles and squares, normal individuals; black circles and squares indicate affected individuals; bottom filed circles and squares indicate heterozygotes. Abbreviations: M, mutated allele; N, normal allele. B Homozygosity mapping shows that the two affected individuals (bottom two samples in black square box) are homozygous (homozygosity shown as bookmarks) in the region bordered by rs2865147 (77,954,733 bp) and rs17472783 (131,309,534 bp) on chromosome 4. Two unaffected individuals (top two samples) are not homozygous in this region. C Sequence chromatograms of variant in the PRDM8 gene
Genome-wide linkage analysis
DNA was isolated according to the salting-out method. Genome-wide single-nucleotide polymorphism (SNP) genotyping was performed on DNA samples of four individuals of the HSP100 family using HumanCytoSNP-12v2-1_L BeadChips (Illumina; www.illumina.com). The individuals included two affected siblings and their unaffected parents (Fig. 1A). This chip includes ~ 300,000 SNPs and can also detect cytogenetic abnormalities. Data were analyzed using GenomeStudio_Genotyping_Module_V1.0 (Illumina). SNPs with Mendelian error and non-call SNPs were removed. Homozygous regions common to affected siblings with a minimum physical length of 1 Mb and absent in parents were sought.
Exome sequencing
Exome sequencing was performed on the DNA of the proband (HSP100-IV2) by Illumina HiSeq 2500 system (Illumina). Sequence alignment and variant calling were performed against human reference genome UCSC NCBI37/hg19. Preliminary filtering was carried out to identify all homozygous/compound heterozygous/X-linked variants. Then, variants that did not affect amino acid change or splicing were filtered out. Subsequently, SNPs with a reported minimal allele frequency (MAF) more than 0.01 in the 1000 Genomes database (www.1000genomes.org), the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), the Genome Aggregation Database (http://genomad.broadinstitute.org/), the Healthy Exomes database (https://www.alzforum.org/exomes/hex), and Iranome database (http://iranome.com/), or observed in in-house exome data of 100 unrelated Iranians affected with non-neurological diseases, were removed. The remaining variants were examined to identify those within any of the known HSP- or other neurodegenerative disease-causing genes, including amyotrophic lateral sclerosis (ALS), neurodegenerations with brain iron accumulation (NBIA), Parkinson disease (PD), and myoclonus epilepsy and neuropathies (Supplementary Table S1). Finally, variants located within the homozygous region or consistent with X-linked inheritance were considered. The remaining variants were also evaluated based on the American College of Medical Genetics (ACMG) criteria [26].
Screening of the candidate variants
Nine candidate disease-causing variants, c.A12193G:p.I4065V and c.A13039G:p.I4347V in SACS, c.A1195G:p.K399E in FRMPD4, c.G1694A:p.R565H in DRP2, c.G109A:p.D37N in OTUD6A, c.G278A:p.R93Q in GLRA4, c.G491T:p.G164V in CXXC4, c.C1003T:p.R335W in OCLN, and c.C689G:p.A230G in PRDM8, were amplified from the DNA of the proband by polymerase chain reaction (PCR). The PCR products were sequenced using the Sanger method. Confirmed variants in the proband were screened in family members for co-segregation analysis with the disease status by direct sequencing.
Furthermore, the variant of PRDM8 was also screened in the 200 additional Iranian control individuals by amplification refractory mutation system-polymerase chain reaction (ARMS-PCR) and in the 100 individual’s in-house exomes data on Iranian peoples.
Skin biopsy
For histopathological evaluation of Lafora bodies, axillary skin punch biopsy (0.4 × 0.3 × 0.3 cm) was done for both affected siblings. Periodic acid-Schiff (PAS) staining with and without diastase treatment, an enzyme that digests the glycogen, was performed.
Results
Results of clinical presentations
HSP100-IV2 (proband)
He presented with a delay in motor development and started walking after 3 years old. After physiotherapy, he could walk on his toes by 5 years old. He studied only to the first grade in school and began exhibiting school difficulties in the second grade. Then he developed progressive lower-limb spasticity and weakness, hand tremor, and dysarthria later on followed by ataxia and urinary incontinence. He became wheelchair-bound by the age of 10 years. At present, he is 13 years old and has not reported any history of seizures.
On neurologic examination, he was fully oriented to time, place, and persons and followed the orders. Due to illiteracy, he was not cooperative enough to do a cognitive test such as Montreal Cognitive Assessment (MoCA). He had scanning speech and a mild jerky tremor in the head. He had abnormal facial movements with persistent contraction of the brow and eyelids as well as the lower face. There was additional tremulousness of the facial muscles and jaw as well as possible slightly rhythmic movements of the lower face. Electromyographic study of facial muscles confirmed tremor with a frequency of 3–4 Hz. Upper limbs showed finger to nose dysmetria and dysdiadocokinesia.
He had a mixture of tremor and athetoid movements of the fingers and dystonic posturing in his hands. He was slow and stiff in upper extremities and had severe spasticity of lower extremities with equinovarus deformity of the feet. Deep tendon reflexes were increased in the upper and lower extremities with bilateral Babinski signs. Gait required bilateral assistance with severe spasticity and prominent scissoring of his legs (Video 1).
Brain magnetic resonance imaging (MRI) was entirely normal without evidence of cerebellar atrophy. Electromyography/nerve conduction studies (EMG/NCS) were normal with no evidence of myopathy or polyneuropathy. Electroencephalography (EEG) showed bursts of generalized sharp waves (Supplementary Figure S1).
HSP100-IV1
The second sibling presented similar features to his brother. He also displayed motor developmental delay and started walking after 2 years old. He became wheelchair-bound by the age of 13 and could walk on his toes before that. He studied to the 6th grade in school and then he revealed school difficulties. His IQ test was borderline. Gradually, he developed spasticity, progressive weakness of the lower limbs and severe ataxia and dysarthria, foot drop and deformity, hand tremor, and urinary incontinence. Like his affected sibling, he did not exhibit any generalized seizures to date.
Examination revealed scanning speech and head tremor (titubation). There was abnormal contraction (probably dystonia) of upper and lower facial muscles associated with the tremor of the jaw and facial muscles. There was a mixture of abnormal movements in his upper limbs with tremor, athetoid movements, dystonic posturing, and jerks in the arms and fingers when he outstretched the hands (probably myoclonus). There was dysmetria on the finger to nose test and dysdiadocokinesia. He had severe spasticity of lower extremities with equinovarus deformity of feet. Deep tendon reflexes were increased in upper and lower extremities with bilateral sustained ankle clonus. Plantar reflexes were extensor on both sides (Babinski sign). He was not able to walk even with bilateral assistance (Video 2).
Like his brother, his brain MRI and EMG/NCS were normal. EEG showed bilateral synchronous sharp waves in frontal areas but no generalized slowness (Supplementary Figure S2).
Results of genetic analyses
The GenomeStudio Homozygosity Detector tool of the GenomeStudio program identified nine regions on chromosomes 3, 4, 5, 6, 17, 18, and 22 that were homozygous for the two affected siblings and were not homozygous in the unaffected parents (Fig. 1B). The proximal and distal markers of these regions and their length have been shown in Supplementary Table S2. No copy number variations (CNVs) were detected.
After preliminary filtering of WES data discussed above, 26 variants with MAF < 0.01 in public databases were identified (Supplementary Table S3). Respectively, 3, 4, and 2 of these variants had been located in homozygous regions, X-chromosome, or were in the compound heterozygous state (Supplementary Table S3).
All nine candidate variants were screened in the family members and only a novel missense variant, c.C689G:p.A230G, in the PRDM8 gene co-segregated with disease status in the family. Both affected members carried homozygous mutations, whereas six unaffected individuals carried at least one normal allele, consistent with autosomal recessive inheritance (Fig. 1C). The candidate variant in PRDM8 was observed only in gnomAD v2.1.1 in a heterozygous state (minor allele frequency; MAF = 0.000021). It was not present in other exome/genome databases mentioned in the “Methods” section and especially in the IRANOME database that reports exome sequences data of 800 healthy Iranian individuals. Also, the variant was not detected in the 300 additional Iranian control individuals. None of them had the variant in either the heterozygous or homozygous states. It was anticipated as a “Likely pathogenic” variant by adjusted ACMG criteria and Varsome (https://varsome.com/).
A search of clinical databases showed the variant has been submitted to ClinVar database with accession ID: SCV001372980.2 on January 07, 2021. So, based on all the below evidence, the PRDM8 variant was considered a candidate disease-causing variant in the HSP100 family (Supplementary Table S3); (i) it is located in the linked/homozygosity region, (ii) it is located in a gene definitively known to cause the disease and co-segregated with the disease in multiple affected individuals of the family, (iii) it was absent from controls or its homozygous allele count was less than 3 or its allele frequency was less than 0.0001 [22], (iv) evolutionarily, Ala230 was a conserved amino acid among mammalian orthologue proteins, (v) there was the same variant in the ClinVar, and (vi) there were similarities between the clinical features of our cases and Pakistani cases with another mutation in the PRDM8 gene.
Pathological findings
Periodic acid-Schiff (PAS) staining of sections of the axillary skin biopsies of the proband and his brother, with and without diastase treatment, showed no evidence of skin Lafora bodies (Fig. 2A and B). The family declined consent for a muscle biopsy, and we were unable to determine the existence or lack of Lafora bodies in the skeletal muscle.

Skin biopsies of axillary apocrine glands with periodic acid-Schiff (PAS) staining. There is no evidence of Lafora bodies. A HSP100-IV2; B HSP100-IV1
Discussion
Approximately, 93% of all families affected with Lafora disease have carried mutations in the EPM2A or EPM2B gene [19, 27] that suggests the involvement of other unknown genes in the remaining (7%) families. Turnbull et al. in 2012 identified a mutation in a novel gene, PRDM8, as the cause of early-onset Lafora disease in a Pakistani family [12]. A decade from the first report and in the present study, we have identified a second probable disease-causing variant in the PRDM8 gene in an Iranian family suspected to be the complicated form of HSP. Our experience expanded the phenotypic spectrum of mutations in the PRDM8-related disorders. There were intra- and inter-familial similarities and differences in these families. The Pakistani cases were older than Iranian patients which may explain these clinical differences. While dystonia and equinovarus were only observed in the Iranian patients, behavioral/neuropsychiatric disorders and epilepsy were only detected in the Pakistani cases. Ataxia, spasticity, dysarthria, and mental impairment or school difficulties were manifested in the affected individuals of both families (Table 1). Myoclonus was the first symptom in two Pakistani siblings, but the third case presented myoclonus at age 21 years [12], while it was only a mild feature in the older Iranian brother. Altogether, as mentioned previously, myoclonus and seizures are less severe in the early-onset LD than other subtypes of LD. Thus, these manifestations may be mild or present later as the disease progresses. As specified in the definition of the early-onset LD, (i) its age at onset is about 5 years and (ii) phenotypic presentations have some differences with other subtypes of progressive myoclonus epilepsies; visual impairment is not observed in early-onset LD however; dysarthria is an important and severe symptom of the disease [12], just as observed in our patients. (iii) The course of disease in early-onset LD is more prolonged than classic LD, and patients can be alive into the fourth decade of life, while most patients affected by classic LD are dead by the age of 30 years [3, 12]. (v) Also, it seems the skin Lafora bodies are not detected in the early-onset LD than classic LD. Like the Pakistani family [12], Lafora bodies were not detected in skin biopsies of the affected members of the Iranian family (Fig. 2A and B). However, the skeletal muscle biopsy of two Pakistani patients showed numerous Lafora bodies. Unfortunately, our family declined muscle biopsies. Although we have not been able to confirm the presence of Lafora bodies in skeletal muscle, however, based on the phenotypic presentations and results of genetic analysis, we suggest that our patients may be affected by early-onset LD, or, given our evidence for an expanded phenotypic spectrum; we propose that they may be affected by a PRDM8-related disorder. It seems reasonable that other PRDM8-related patients should be assessed for the presence or absence of Lafora bodies in their skin. Also, in the advanced stages of the disease, the formation of Lafora in their skin should be re-checked.
PRDM8 protein contains a nuclear localization signal (NLS) suggesting that it might be a nuclear protein. It interacts with laforin phosphatase and malin ubiquitin E3 ligase, translocates them from the cytoplasm to the nucleus, and forms distinctive punctate foci [12]. The mutant PRDM8 (F261L-PRDM8) over-sequesters laforin and malin in the nucleus much more than the wild type and leads to deficiencies of these proteins in the cytoplasm [12]. Laforin and malin normally contribute to glycogen synthesis, and it is suggested that their deficiencies can lead to the formation of insoluble polyglucosans instead of glycogen [12]. Formation and accumulation of polyglucosans in neuronal tissue may interfere with normal cellular functions and contribute to neurodegeneration, and finally clinical manifestations of the disease [4]. In support of these findings, a knockout mouse model of PRDM8 manifests abnormal axon guidance [28]. However, the role of Lafora bodies in early-onset LD remains uncertain and requires clarification in future studies. Further studies on PRDM8 will hopefully uncover its possible role in regulation of glycogen construction and lead to better recognition of the biological pathways and functions of this gene.
Evolutionarily, PRDM8 has been detected only in the vertebrates and alanine at position 230 is completely conserved in human PRDM8 and mammalian orthologue proteins (Fig. 3). The c.C689G:p.A230G variant is located in exon 4 of the PRDM8 gene (NM_001099403) and close to the previously reported variant, c.T781C:p.F261L, in the Pakistani family. So, we expect the variant can cause similar phenotypic symptoms.

A PRDM8 protein domain: five conserved domains including three zinc finger domains (CH2 and CH2-like), a nuclear localization signal (NLS), and a Proline-rich (PR) domain. Arrow and star indicate positions of the Iranian and Pakistani PRDM8 variants, respectively. Ala230 is part of a conserved block of amino acids across mammalians (indicates with underline). B Evolutionarily conservation of alanine at position 230 (in Iranian family) and phenylalanine at position 261 (in Pakistani family) of PRDM8 in human and mammalian orthologue proteins
Both reported families with mutations in PRDM8 have originated from the Middle East, so additional cases of the disease may probably be found in this region. Given that about 7% of all families affected with Lafora disease do not carry a mutation in EPM2A or EPM2B gene, we suggest that these families should be screened for PRDM8 mutations. Screening of PRDM8 in the large cohort of LD cases without mutations in the classic genes may result in identification of other PRDM8-related cases and expand the spectrum of mutations in this gene. PRDM8 mutations are not likely the cause of classic LD but those should be considered in patients with progressive myoclonus, dysarthria, and ataxia, or even cases with other neurodegenerative disorders especially in the Middle East.
Due to the false results, a skin biopsy should not be relied on for the diagnosis. Lafora bodies on muscle biopsy are strongly supportive, although the sensitivity of this finding for a diagnosis of early-onset LD is not known and the muscle biopsy is an invasive method. Therefore, the disease should be confirmed by the genetic analysis as the “gold standard” method [1].
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
References
Jansen AC, Andermann E (2019) Progressive myoclonus epilepsy, Lafora type, GeneReviews®[Internet]. University of Washington, Seattle
Al Mufargi Y, Qureshi A, Al Asmi A (2020) Lafora disease: report of a rare entity. Cureus 12(1):e6793
Minassian BA (2001) Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol 25(1):21–29
Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B, Criado-García O, Fernandez-Sanchez E, Medraño-Fernández I, Domínguez J (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10(11):1407–1413
Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BA (2018) Lafora disease—from pathogenesis to treatment strategies. Nat Rev Neurol 14(10):606–617
Nitschke F, Ahonen SJ, Nitschke S, Mitra S, Minassian BAJNRN (2018) Lafora disease—from pathogenesis to treatment strategies. Nat Rev Neurol 14(10):606–617
Delgado-Escueta A, Ganesh S, Yamakawa K (2001) Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet 106(2):129–138
Brewer MK, Putaux J-L, Rondon A, Uittenbogaard A, Sullivan MA, Gentry MS (2020) Polyglucosan body structure in Lafora disease. Carbohydr Polym 240:116260
Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS (2012) Glycogen and its metabolism: some new developments and old themes. Biochem J 441(3):763–787
Delgado-Escueta AV (2007) Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep 7(5):428–433
Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, DePaoli-Roach AA, Roach PJ (2008) Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem 283(49):33816–33825
Turnbull J, Girard J-M, Lohi H, Chan EM, Wang P, Tiberia E, Omer S, Ahmed M, Bennett C, Chakrabarty A (2012) Early-onset Lafora body disease. Brain 135(9):2684–2698
Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJJNG (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet 35(2):125–127
Turnbull J, DePaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, Piliguian M, Roach PJ, Wang P, Ackerley CA (2011) PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet 7(4):e1002037
Serratosa JM, Gómez-Garre P, Gallardo ME, Anta B, De Bernabé DB-V, Lindhout D, Augustijn PB, Tassinari CA, Michelucci R, Malafosse AJHMG (1999) A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet 8(2):345–352
Chan E, Omer S, Ahmed M, Bridges L, Bennett C, Scherer S, Minassian BJN (2004) Progressive myoclonus epilepsy with polyglucosans (Lafora disease): evidence for a third locus. Neurology 63(3):565–567
Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 11(11):1251–1262
Nitschke F, Sullivan MA, Wang P, Zhao X, Chown EE, Perri AM, Israelian L, Juana-López L, Bovolenta P, Rodríguez de Córdoba S (2017) Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. EMBO Mol Med 9(7):906–917
Singh S, Satishchandra P, Shankar SK, Ganesh S (2008) Lafora disease in the Indian population: EPM2A and NHLRC1 gene mutations and their impact on subcellular localization of laforin and malin. Hum Mutat 29(6):E1–E12
van de Peppel J, Holstege FC (2005) Multifunctional genes. Mol Syst Biol 1(1):2005.0003
Inoue M, Kuroda T, Honda A, Komabayashi-Suzuki M, Komai T, Shinkai Y, Mizutani K-I (2014) Prdm8 regulates the morphological transition at multipolar phase during neocortical development. PLoS One 9(1):e86356
Jung CC, Atan D, Ng D, Ploder L, Ross SE, Klein M, Birch DG, Diez E, McInnes RR (2015) Transcription factor PRDM8 is required for rod bipolar and type 2 OFF-cone bipolar cell survival and amacrine subtype identity. Proc Natl Acad Sci 112(23):E3010–E3019
Orouji E, Peitsch WK, Orouji A, Houben R, Utikal J (2020) Unique role of histone methyltransferase PRDM8 in the tumorigenesis of virus-negative Merkel cell carcinoma. Cancers 12(4):1057
Park J, Kwon SO, Kim S-H, Kim SJ, Koh EJ, Won S, Kim WJ, Hwang SY (2020) Methylation quantitative trait loci analysis in Korean exposome study. Mol Cell Toxicol 16:175–183
Neidhart M, Pajak A, Laskari K, Riksen NP, Joosten LA, Netea MG, Lutgens E, Stroes ES, Ciurea A, Distler O (2019) Oligomeric S100A4 is associated with monocyte innate immune memory and bypass of tolerance to subsequent stimulation with lipopolysaccharides. Front Immunol 10:791
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector EJGIM (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423
Ferlazzo E, Canafoglia L, Michelucci R, Gambardella A, Gennaro E, Pasini E, Riguzzi P, Plasmati R, Volpi L, Labate A (2014) Mild L afora disease: clinical, neurophysiologic, and genetic findings. Epilepsia 55(12):e129–e133
Ross SE, McCord AE, Jung C, Atan D, Mok SI, Hemberg M, Kim T-K, Salogiannis J, Hu L, Cohen SJN (2012) Bhlhb5 and Prdm8 form a repressor complex involved in neuronal circuit assembly. Neuron 73(2):292–303
Funding
Genetic Research Center, University of Social Welfare and Rehabilitation Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
All participants, after being informed of the nature of the research, consented to participate. This research was performed in accordance with the Declaration of Helsinki and with approval of the ethics board of the University of Social Welfare and Rehabilitation Sciences.
Conflict of interest
None.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file6 (AVI 13667 KB)
Supplementary file7 (AVI 6323 KB)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Davarzani, A., Shahrokhi, A., Hashemi, S.S. et al. The second family affected with a PRDM8-related disease. Neurol Sci 43, 3847–3855 (2022). https://doi.org/10.1007/s10072-021-05815-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-021-05815-w