
Abstract
Hereditary spastic paraplegia (HSP) includes a number of inherited disorders which are characterized by stiffness in the lower extremities and progressive gait disturbance. Mutations in terms of spastic gait genes (SPGs) are responsible for occurrence of different types of HPS with autosomal recessive, X-linked recessive, and autosomal dominant modes of inheritance. In the current case report, we identified a mutation in SPG11 gene in a female patient with progressive stiffness of lower extremities and atrophy of corpus callosum and the “lynx ear” sign in brain MRI. Whole exome sequencing (WES) revealed a homozygote frameshift deletion variant in SPG11 gene (NM001160227: exon 28: c.4746delT, p.N1583Tfs*23). This variant is a null variant classified as a pathogenic variant (PVS1) according to ACMG standards and guidelines. The frequency of this variant in 1000G, ExAC, and Iranome databases was 0. This study shows the role of WES in the identification of disease-causing mutations in a disease such as HSP which can be caused by diverse mutations in several genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary spastic paraplegia (HSP) includes a number of inherited disorders which are characterized by progressive gait disturbance, stiffness, and contraction in the lower extremities (Fink 2003). Other associated signs are brisk reflexes, extensor plantar reflexes, muscle weakness, and urinary urgency (Depienne et al. 2007). Being classified as an upper motor neuron disease, it is caused by degeneration of neurons in the corticospinal tracts (Depienne et al. 2007). Several spastic gait genes (SPGs) are responsible for occurrence of different types of HPS with autosomal recessive, X-linked recessive and autosomal dominant modes of inheritance. However, molecular studies have indicated clustering of HPS-related genes in a limited number of cellular processes such as organelles modeling and intracellular membrane trafficking (Blackstone 2012). Age of disease onset is extremely wide ranging from infancy to elderly. When disease has been manifested during the teenage years or afterward, the progressive course of spastic gait may lead to eventual need for mobility aids. However, when symptoms initiate in the late infancy or early childhood, functional worsening is not expected (Fink 2003). More than 70 genetic loci have been recognized for HSP up to now, and the majority of these genes have been cloned (Giudice et al. 2014). Among the identified genes, those responsible for mitochondrial metabolism, endosomal and trans-Golgi transport, and axonal transmission have been recognized (Crosby and Proukakis 2002). In the current case report, we explain the clinical features and the identified mutation in an Iranian patient with HSP.
Patient and Methods
The patient was a 23-year Iranian female born to a healthy first-cousin parent. She had a healthy younger sib. She was referred to neurology section of Imam Hossein Hospital, Tehran, Iran, for assessment of lower extremity paresis. No similar case has been reported in the family member. She reported this symptom from 4 years before. She was born with normal vaginal delivery. There was no remarkable prenatal history. Growth and development were completely normal. Cranial nerve examination revealed no abnormal sign. Tonicity of upper extremities was normal. Lower extremities were spastic. Deep tendon reflexes were normal except for extensor plantar reflexes. There was no sign of cognitive deficiency and peripheral neuropathy. No other sign was detected in complete physical examination. Brain MRI showed atrophy of corpus callosum (Fig. 1) and the “lynx ear” sign (Fig. 2).

Brain MRI showing atrophy of corpus callosum

Brain MRI of the patient showing ears of the lynx sign
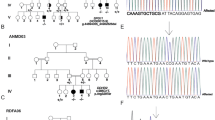
DNA was extracted from peripheral blood of patient using salting-out method. The extracted DNA was sent for whole exome sequencing (WES). WES identified a homozygote frameshift deletion variant in SPG11 gene (NM001160227: exon 28: c.4746delT, p.N1583Tfs*23). This variant is a null variant classified as a pathogenic variant (PVS1) according to ACMG standards and guidelines. The frequency of this variant in 1000G, ExAC, and Iranome databases was 0. Parent had the mutation in heterozygote state.
Discussion
In the current study, we identified a homozygote mutation in SPG11 in a patient with spastic paraplegia. SPG11 mutations have been recognized as a frequent molecular mechanism for autosomal recessive HSP (Stevanin et al. 2007a). SPG11 gene is located on 15q21.1 and encodes the spatacsin, a ubiquitously expressed protein in the nervous system (Stevanin et al. 2006). SPG11-associated HSP cases are classically manifested by spasticity, cognitive deficiency, and peripheral neuropathy (Stevanin et al. 2007a; Siri et al. 2010). The case presented in the current study had no sign of cognitive deficiency and peripheral neuropathy. However, she had the characteristic signs of corpus callosum atrophy and periventricular white matter changes. The association between HSP and thin corpus callosum has been firstly noted by Nakamura et al. in their clinical assessment of 2 families with autosomal recessive HSP (Nakamura et al. 1995). The cases presented by Nakamura et al. had mild to moderate intellectual disability (Nakamura et al. 1995), yet the current case had normal intelligence. The thin corpus callosum has also been reported in Italian HSP cases presented by Casali et al. (Casali et al. 2004). The present case differs from the Italian cohort in terms of later manifestation of disorder and normal intelligence (Casali et al. 2004). In a cohort of German HSP patients with genetically confirmed mutations in SPG11, dysarthria and mental impairment were reported in the majority of patients (Hehr et al. 2007). Nevertheless, the present case had normal speech. Although the clinical manifestations of SPG11 mutations might be different, this gene is regarded as the most commonly accountable gene for autosomal recessive HSP with thin corpus callosum. However, as Stevanin et al. could not detect SPG11 mutation in a family with these clinical features, it seems that at least one other gene responsible for autosomal recessive HSP with thin corpus callosum remains to be identified (Stevanin et al. 2007b).
The patient had the “ears of the lynx” sign in her brain MRI. This sign denotes the unusual T2/FLAIR-tapered hyperintensity at the tip of the frontal horn of the lateral ventricles which looks like the clumps of hair on the top of the ears of a lynx. Pascual et al. have recently shown the close association between this MRI sign and mutations in SPG11 and SPG15 genes (Pascual et al. 2019). However, this sign is not limited to HSP, as Pacheco et al. have shown this sign in a patient with a chronic form of Marchiafava-Bignami disease (Pacheco et al. 2014).
The present case had a frameshift mutation in exon 28. Southgate et al. have identified a c.4846C > T (p.Q1616X) nonsense mutation in exon 28 in a Pakistani family with similar features of HSP and thin corpus callosum (Southgate et al. 2010). Although few patients with HSP and thin corpus callosum have mutations in exons 18–29 (Southgate et al. 2010), this region of gene encodes the central part of the spatacsin protein which is predicted to have potential structural significance (Stevanin et al. 2007b). SPG11 has 40 exons and the identified mutations have been located in numerous exons with no preferential mutation hot spot (Paisan-Ruiz et al. 2008). Therefore, WES is the preferred method for identification of disease-causing mutations in suspected cases.
It is worth mentioning that in addition to the typical HSP, SPG11 mutations have been associated with a number of neurological conditions such as juvenile-onset parkinsonism (Paisán-Ruiz et al. 2010; Anheim et al. 2009) and dystonia (Yoon et al. 2013; Wijemanne et al. 2015). These phenotypes rarely detected in association with other SPG genes (Giudice et al. 2014). Thus, assessment of phenotypes associated with each mutation would help in elaboration of genotype-phenotype correlation and predication of disease course in future cases. Therefore, the main finding of this study is the demonstration of a distinct phenotype associated with a novel mutation in SPG11. The limitation of our study was lack of in vitro assessment of functional consequences of this nucleotide change.
References
Anheim M, Lagier-Tourenne C, Stevanin G, Fleury M, Durr A, Namer IJ, Denora P, Brice A, Mandel JL, Koenig M (2009) SPG11 spastic paraplegia. J Neurol 256:104–108
Blackstone C (2012) Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 35:25–47
Casali C, Valente E, Bertini E, Montagna G, Criscuolo C, De Michele G, Villanova M, Damiano M, Pierallini A, Brancati F (2004) Clinical and genetic studies in hereditary spastic paraplegia with thin corpus callosum. Neurology 62:262–268
Crosby AH, Proukakis C (2002) Is the transportation highway the right road for hereditary spastic paraplegia? Am J Hum Genet 71:1009–1016
Depienne C, Stevanin G, Brice A, Durr A (2007) Hereditary spastic paraplegias: an update. Curr Opin Neurol 20:674–680
Fink JK (2003) The hereditary spastic paraplegias: nine genes and counting. Arch Neurol 60:1045–1049
Giudice TL, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A (2014) Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 261:518–539
Hehr U, Bauer P, Winner B, Schule R, Olmez A, Koehler W, Uyanik G, Engel A, Lenz D, Seibel A (2007) Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann Neurol 62:656–665
Nakamura A, Izumi K, Umehara F, Kuriyama M, Hokezu Y, Nakagawa M, Shimmyozu K, Izumo S, Osame M (1995) Familial spastic paraplegia with mental impairment and thin corpus callosum. J Neurol Sci 131:35–42
Pacheco FT, Rego MM, Do Rego JIM, Da Rocha AJ (2014) “Ears of the Lynx” sign in a Marchiafava–Bignami patient: structural basis and fiber-tracking DTI contribution to the understanding of this imaging abnormality. J Neuroimaging 24:205–207
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70:1384–1389
Paisán-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M (2010) Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 25:1791–1800
Pascual B, De Bot S, Daniels M, França M, Toro C, Riverol M, Hedera P, Bassi M, Bresolin N, Van De Warrenburg B (2019) “Ears of the Lynx” MRI sign is associated with SPG11 and SPG15 hereditary spastic paraplegia. Am J Neuroradiol 40:199–203
Siri L, Battaglia F, Tessa A, Rossi A, Rocco MD, Facchinetti S, Mascaretti M, Santorelli F, Veneselli E, Biancheri R (2010) Cognitive profile in spastic paraplegia with thin corpus callosum and mutations in SPG11. Neuropediatrics 41:35–38
Southgate L, Dafou D, Hoyle J, Li N, Kinning E, Critchley P, NÉmeth AH, Talbot K, Bindu PS, Sinha S (2010) Novel SPG11 mutations in Asian kindreds and disruption of spatacsin function in the zebrafish. Neurogenetics 11:379–389
Stevanin G, Montagna G, Azzedine H, Valente EM, Durr A, Scarano V, Bouslam N, Cassandrini D, Denora PS, Criscuolo C (2006) Spastic paraplegia with thin corpus callosum: description of 20 new families, refinement of the SPG11 locus, candidate gene analysis and evidence of genetic heterogeneity. Neurogenetics 7:149–156
Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, Rosa AL, Lerer I, Hamri A, Alegria P (2007a) Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 131:772–784
Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, Martin E, Ouvrard-Hernandez A-M, Tessa A, Bouslam N (2007b) Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 39:366
Wijemanne S, Shulman JM, Jimenez-Shahed J, Curry D, Jankovic J (2015) SPG 11 mutations associated with a complex phenotype resembling dopa-responsive dystonia. Mov Disord Clin Pract 2:149–154
Yoon G, Baskin B, Tarnopolsky M, Boycott K, Geraghty M, Sell E, Goobie S, Meschino W, Banwell B, Ray P (2013) Autosomal recessive hereditary spastic paraplegia—clinical and genetic characteristics of a well-defined cohort. Neurogenetics 14:181–188
Conflict of Interest
The authors declare they have no conflict of interest.
Funding
This study was financially supported by Shahid Beheshti University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sayad, A., Akbari, M.T., Hesami, O. et al. Identification of a Mutation in SPG11 in an Iranian Patient with Spastic Paraplegia and Ears of the Lynx Sign. J Mol Neurosci 70, 959–961 (2020). https://doi.org/10.1007/s12031-020-01501-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01501-2