
Abstract
Background
Environmental exposure and genotype variation influence DNA methylation. Studies on the effects of genotype variation were performed mainly on European ancestries. We analyzed the genetic effects on cord blood methylation of Koreans.
Methods
As part of the Korean Exposome study project, DNA was extracted from 192 cord blood samples for analysis. Cord blood samples were genotyped via Asian Precision Medicine Research Array analysis and methylation was measured using the Methylation EPIC Beadchip kits. The associations between genotypes and CpG methylation were analyzed with matrix eQTL.
Results
Conditional analysis revealed 34,425 methylation quantitative trait loci (mQTLs), and trans-mQTLs constituted 7.2% of all the associated CpG sites. About 80% of the total trans-associations were trans-chromosomal and the related SNPs were concentrated on chromosome 19. According to the results of DAVID, cis-mQTL-related SNPs resulting in amino acid substitutions were related to signal peptides or glycosylation.
Conclusion
We identified genotype variations associated with DNA methylation in the cord blood obtained from Koreans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent studies have shown that environmental exposures and genotype variation affect human traits and diseases via epigenetic mechanisms (Feinberg 2018). Epigenetics is a study of the changes in gene function in the absence of gene mutations, and involves transmission of specific acquired traits to the offspring regardless of the gene sequence. DNA methylation is one of the important epigenetic mechanisms that regulate gene expression. It entails a typical widespread genetic variation that alters gene function in the absence of a mutation, for transmission to offspring, and is associated with disease development.
However, the exact mechanisms underlying pathogenesis due to altered DNA methylation have yet to be elucidated. Recent studies investigating the relationship between DNA methylation and disease or human traits explain the underlying possible mechanisms (Stricker et al. 2016).
Cord blood DNA methylation studies have successfully identified CpG loci associated with in utero environmental exposure or related to health outcomes (Joubert et al. 2016; Küpers et al. 2019). Systemic analysis of genetic effects on methylation during human life span suggests that the genetic component of methylation is causally related to complex traits (Gaunt et al. 2016).
Until now, studies investigating the effect of genotype on methylation in different tissue types have been mainly based on European ancestries (Shi et al. 2014). Because ethnic groups show differences in genetic variation as well as environmental exposure, studies are needed to analyze the genetic effects on DNA methylation in different ethnicity. In this study, we analyzed the genetic effects on cord blood methylation in Koreans.
Methods
Study samples
As part of the Korean Exposome study project, 192 cord blood samples were selected for analysis. This study received ethical approval from the Kangwon National University Hospital IRB (B-2017-11-006).
Genotype data
Cord blood was genotyped with the Asian Precision Medicine Research Array (APMRA, Affymetrix). DNA samples were hybridized to an array in the GeneTitan MC Instrument (Affymetrix, CA, USA) according to the GeneTitan® Multichannel Instrument User’s Manual, using the Axiom APMRA 96-ARRAY. After ligation, the arrays were stained and imaged on the GeneTitan MC Instrument (Affymetrix, CA, USA).
The quality control of genotype data was conducted with plink v1.90b3.44 (Chang et al. 2015) and WISARD 1.3.3. Samples were filtered according to the following standards: (1) male subjects with X chromosome inbreeding coefficient less than 0.8 or female subjects with X chromosome inbreeding coefficient larger than 0.2, (2) subjects with the genotype missing rate greater than 5%, (3) related subjects based on the identity-by-state (IBS) matrix value larger than 0.8, and (4) outliers from MDS plots drawn with three principal component (PC) scores. The filtering of variants was performed with the missing rate (> 5%), the Hardy–Weinberg equilibrium (HWE) p value (< 10−5), and the minor allele frequency (MAF, < 5%).
Missing genotypes in the filtered data were filled in the pre-phasing step with SHAPEIT v2.r837 (Delaneau et al. 2012). The phased genotype data were then imputed to the whole-genome sequencing data level with IMPUTE 2.3.2 with 1000 Genome Project phase 3 reference data containing 2504 subjects (Howie et al. 2009, 2011, 2012). The screen size of 3 Mbp and the buffer size of 1 Mbp were used. The same procedure of variant filtering was used after the imputation, and the results with information score not less than 0.9 were retained.
Methylation data
DNA methylation was measured using MethylationEPIC BeadChip kits (Illumina, CA, USA) according to the manufacturer’s instructions. The BeadChips were imaged via Illumina iScan System (Illumina, CA, USA) using the manufacturer’s standard protocol.
Sample quality control of methylation data was conducted with an R package meffil (Min et al. 2018). The CpG sites were filtered with R packages ENmix (Xu et al. 2015) and minfi (Aryee et al. 2014). The outliers were detected based on the difference in X–Y chromosome probe intensity, and defective subjects were filtered out according to the medians of methylated and unmethylated signals, control probe intensities, or the discordance with the genotype data. Also, CpG sites were removed (1) if more than 5% of the samples were flagged as bad (detection p value > 10−6 or bead number < 3), (2) or if the corresponding probe contains an SNP at the CpG interrogation or at the single-nucleotide extension.
The raw intensity data were background corrected with ENmix (Xu et al. 2015), with the background normal distribution parameters estimated with out-of-band values. The corrected intensities were subjected to beta-mixture quantile normalization method (Teschendorff et al. 2012). Batch effects were adjusted by ComBat (Johnson et al. 2007). The resulting beta values were inverse normal transformed (INT) as the regression models assumed the normality of the data.
Cell composition estimation
From the methylation data, the leukocyte composition for each sample was estimated with Houseman algorithm (Houseman et al. 2012). The Bakulski reference was used as cord blood reference panel for the estimation of CD8+, CD4+, natural killer cells, monocytes, granulocytes, B cells, and nucleated red blood cells (Bakulski et al. 2016). In the following analysis, the granulocytes were eliminated to avoid multi-collinearity, since the variance inflation factor was the largest.
Methylation quantitative trait loci (mQTL) analysis
The associations between genotypes and CpG methylations were analyzed with matrix eQTL (Shabalin 2012), using codes written by Gaunt et al. (2016). The regression analysis was adjusted for maternal age, birth history (0/1), infant sex, leukocyte compositions, and ten genotype PC scores. The missing values in the maternal information were imputed with the average of the values obtained from the other subjects.
Multiple testing was adjusted by Bonferroni correction and the genome-wide significance cut-off was set as 1.672 × 10−14. The results were marked as cis if the SNP and the CpG site were within 1 Mb to each other, and otherwise, the results were designated as trans. The Manhattan plots were drawn using an R package qqman (Turner 2014).
Conditional analysis
To select the most representative variants associated with the methylation of each CpG site, the conditional association analysis was performed with COJO in GCTA64 1.92.0 beta2 (Yang et al. 2011, 2012). The summary statistics derived from the results of mQTL analysis were used, and a stepwise model selection procedure was adopted to select the SNPs. The circos plot was drawn with an R package circlize (Gu et al. 2014).
Functional annotation analysis
To investigate the functional significance of the methylation-related variants, we used DAVID 6.8 (Huang et al. 2008, 2009). From the results of conditional analysis, we annotated each SNP with the predicted genetic or protein consequence based on Ensembl GRCh37. The unique list of annotated genes was used as an input for DAVID. We used two types of lists: the whole list of annotated genes and the list of genes annotated with non-synonymous variations.
Results
Quality control (QC)
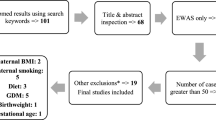
The overall QC flow and the results are shown in Fig. 1. Following QC of genotype data, 1 subject was removed due to the missing rate, 2 subjects were excluded as related subjects, and 2 subjects were considered as outliers from MDS plots. For variants, 6853, 2421, and 392,159 were removed according to the missing rate, the HWE p value, and the MAF, respectively. As a result, 187 subjects and 365,296 variants remained, 354,681 of which were autosomal. Only autosomal variants were used in the following steps.

Flowchart outlining the quality control (QC) steps. The left describes the QC of the genetic data, consisting of subject filtering, variant filtering, and imputation steps. The right describes the QC of the methylation data, composed of subject filtering and site-filtering steps
After the imputation, 10,658,914 variants were added to the data. Among a total of 11,013,595 SNPs, 2,115,146, 45, and 5,124,791 were deleted because of the missing rate, the HWE p value, and the MAF, respectively. Additionally, 91,206 variants with an information score lower than 0.9 were discarded, resulting in a balance of 3,682,407 variants.
QC was performed for methylation data as well. After 4 subjects were detected as outliers and 4 were marked as bad subjects, and 1 of them was both an outlier and a bad subject, the remaining number of subjects was 185. Among CpG sites, 6169 sites with a bad quality in more than 5% of the subjects were discarded. Furthermore, 29,043 SNP-containing probes were excluded, and 812,115 CpG sites only on autosomes were used in the analysis.
Among 192 subjects, 181 common to all of the genotype, methylation, and covariate files were used in the mQTL analysis. These subjects were used in the following analyses.
mQTL analysis
Among 2.991 × 1012 tests (3,682,407 × 812,115), 1,613,688 SNP and CpG site pairs showed significant association, 116,693 (7.23%) of which were trans-mQTLs. For each locus, 57 methylation sites at most were significantly associated. However, each of the CpG sites was associated with a much larger number of mQTLs. For example, the methylation of cg11707556 was significantly related to the genotypes of 3294 SNPs. The results involving the genotype MAF under 0.15 tended to be fewer than the others.
We observed several spots, on chromosome 10 for example, where the SNPs associated with a large number of CpG sites were concentrated. Most of them were due to the cis-mQTLs, while the peak on chromosome 11 (rs7952713) consisted of 44 trans-associations (Fig. 2). Rs7952713 is on the intron of a gene LDLRAD3, which encodes low-density lipoprotein receptor class A domain-containing protein 3. Six SNPs on chromosome 10 showed a cis-mQTL relation with the largest number of 57 CpG sites. The positions of SNPs ranged from 123,207,298 to 123,212,302, which were located upstream of FGFR2. The related CpG sites were located between 122,740,998 and 123,492,885, ranged from upstream of MIR5694 to downstream of FGFR2.

Manhattan plots representing the number of associated CpG sites for each genetic variant. Each point indicates a p value for each variant. Points are ordered by their genomic position, and different chromosomes are indicated by alternating colors of black and gray
Major histocompatibility complex (MHC)-related regions [chr6:28,477,797–33,448,354 in build GRCh37, according to Genome Reference Consortium (GRC)] contained more than 25,000 significant results, with 7114 unique loci and 342 CpG sites. Twelve SNPs around the position 29,643,877–29,648,377, ranging from the body of the gene ZFP57 to the downstream, had the largest number of correlated CpGs of 45, which matched the peak of the Manhattan plot on chromosome 6 (Fig. 2). Among the significant associations with SNPs in the MHC-related regions, 413 (1.61%) were trans-mQTLs. This proportion is much lower than the overall proportion of trans-mQTLs (7.23%). Besides, only 200 (0.79%) among the significant mQTLs with the CpGs in the MHC-related regions were trans-mQTLs.
Interestingly, we could find an SNP on chromosome 16 (rs3809627) had trans-mQTL relationships with 14 CpG sites on chromosome 4 with the positions from 81,048,483 to 81,119,473. The region ranged from the upstream to the body of the gene PRDM8, which encodes a protein belonging to a conserved family of histone methyltransferase. Rs3809627 is on the 5′-UTR of a transcript of TBX6, T-box transcription factor 6.
Conditional analysis
We conducted the conditional analysis, and 34,425 associations (among 1,613,688) with 34,272 unique CpG probes were genome-wide significant after stepwise model selection. Among the 153 sites linked to 2 SNPs, 28 sites carried both cis- and trans-mQTLs. Both CpG sites with cis-mQTL and sites with trans-mQTL showed regional differences in abundance, with lower proportions in the shelf regions (Fig. 3). Compared with the sites carrying cis-mQTL, the CpG sites with trans-mQTL were concentrated on the body of CpG islands. We also found that the abundance varied between genetic locations and the highest proportions except CpG sites without the annotation were inside the body of genes (Fig. 4).

Proportion of CpG sites with cis- (red), trans- (green), and both (blue) mQTLs annotated with the relative positions to UCSC CpG islands. Sites with multiple categories of annotations were counted in duplicate. N_shelf, N_shore, Island, S_shore, and S_shelf indicate shelves 2 kb–4 kb upstream from islands, ~ 2 kb upstream positions flanking islands, body of CpG islands, ~ 2 kb downstream positions from islands, and 2 kb–4 kb downstream from islands respectively

Proportion of CpG sites with cis- (red), trans- (green), or both (blue) mQTLs annotated with genomic locations relative to genes. Sites with multiple annotation categories were counted in duplicate. TSS1500, TSS200, UTR, and ExonBnd indicate 200–1500 nucleotide upstream from transcription start site (TSS), ~ 200 nucleotide upstream from TSS, untranslated region, and exon boundaries, respectively
The circus plot of trans-mQTLs is presented in Fig. 5. About 80% of the total 1508 trans-associations were trans-chromosomal. The related SNPs were concentrated on several regions such as chromosome 19, and the CpG sites were densely located on several areas such as chromosomes 16 and 17.

Circos plot of trans-mQTLs. Sections are separated by chromosomes, marked by the outermost texts, and aligned in the clockwise direction. Values from outside are as follows: − log10 (p value) of each association (dark red points), the density of genetic loci (pink curves), the density of related CpG sites (blue curves), and transassociation between CpG methylations and mQTLs (curved lines)
Functional annotation analysis
According to the results of DAVID, cis-mQTL-related SNPs resulting in amino acid substitutions were related to signal peptides or glycosylation. However, trans-mQTL-related SNPs, regardless of the non-synonymous variation, were highly related to Kruppel-associated box (KRAB) domain, a transcriptional repression domain which is in zinc -finger proteins. Interestingly, SNPs containing both cis- and trans-mQTL were also linked to the KRAB domain. The significance was lost when the SNPs were restricted to the non-synonymous variants.
Discussion
In this study, we identified 34,425 mQTLs based on the genotype and methylation data collected from the cord blood of Koreans. We found that 7.23% of all CpG sites were trans-mQTL, and compared with the sites containing cis-mQTL, CpG sites with trans-mQTL were concentrated on the body of CpG islands. Overall, these conclusions are closely related to the results reported by Gaunt et al. (2016), in which 7% of the mQTLs identified showed trans-effects, and the cis genetic effects on methylation levels increased substantially in the regulatory region.
In this study, approximately 80% of the total 1508 trans-associations were trans-chromosomal, whereas 65% of trans-associations were trans-chromosomal in Gaunt et al. (2016). The related SNPs were concentrated on chromosome 19, which was similar to a previous report (McRae et al. 2018). We also found that the abundance differed between genetic locations and the highest proportions except CpG sites without the annotation were inside the body of genes.
DNA methylation at the genome level is a measurable trait where environmental or genetic variations occur. DNA methylation mediates the effects of environmental and genetic variation on complex diseases (Bell et al. 2011). Cord blood methylation not only affects birth outcome and children’s health outcome, but is also related to health outcome during the course of life (den Dekker et al. 2019). The role of genetic variation in DNA methylation has been demonstrated by studies investigating the heritability using twin pairs and families across various tissues (Boks et al. 2009; Gordon et al. 2012; McRae et al. 2014).
MHC regions, on chromosome 6, are dense of diverse genes, about 40% of which were estimated to have immune system functions (Consortium TMs 1999). In this study, more than 25,000 mQTL relations could be found in the regions. Importantly, several genetic loci nearby ZFP57 had negative correlations with a number of CpG sites that were also annotated with ZFP57 which plays a crucial role in maintenance of DNA methylation status (Mackay et al. 2008; Takahashi et al. 2019; Ferguson-Smith 2011). This may indicate that the methylation status of ZFP57 itself can be regulated by the genotype of ZFP57 or several loci in MHC-related regions, further affecting methylation statuses of several regions. CpG positions whose methylation statuses were correlated with the genotype of loci in MHC regions include MOG and RYR1.
The analyses also found that a locus on the 5′-UTR of TBX6 (chromosome 16) was related to several methylation positions nearby PRDM8. Rs3809627 has been reported to be a potential regulator of the expression of TBX6 (Consortium EP 2012; Wu et al. 2015), and this gene plays an important role in spine development during somitogenesis as a transcription factor (Chen et al. 2016). PRDM8 was found to relocate laforin and malin from cytoplasm to nucleus (Turnbull et al. 2012), possibly affecting the glycogen metabolism in cytoplasm. It may be hypothesized that the altered expression of TBX6 may be related to several glycogen-related phenotypes such as Lafora disease.
The previous meta-analysis by Pregnancy and Childhood Epigenetics (PACE) consortium revealed over 6000 CpG sites of which the methylation statuses in newborn blood were significantly differed by the maternal smoking (Joubert et al. 2016). We found that 398 of the reported sites were found to have significant mQTLs, including the positions annotated with some noticeable genes such as PRDM8 and BHMT2. This result may indicate that the effect of maternal smoking on the infant methylation statuses can be partially amplified or canceled out due to the genotype. For example, minor alleles of SNPs nearby BHMT2 (e.g., rs486580) are positively correlated with the methylation of CpG sites annotated with BHMT2 (e.g., cg08328513), and the maternal smoking may further increase the methylation amount of the sites (Joubert et al. 2016).
Our study is significant in two aspects. First, previous studies mostly focused on non-Hispanic whites, and results derived from Asians have been very limited. Genotypes vary by the population or ancestry, and their effects on the phenotype can be different by the population (Carlson et al. 2013). Therefore, our association results between DNA methylations and the genotypes may provide biological insights applicable to the Asian subjects, and can be utilized in later multi-omics studies of cord blood from similar population (Koh and Hwang 2019). Second, we used the Illumina EPIC array with the wider coverage to analyze DNA methylation. This EPIC array covers over 850,000 methylation sites and can reveal further methylation statuses of CpG sites not covered with Illumina Infinium HumanMethylation450. Therefore, our findings may provide some clue of novel biological pathways explaining the relationships between genotypes, DNA methylations, and the phenotypes.
A significant amount of research has been conducted to determine the role of genetic component in methylation and its effects on disease. The mQTL is related to a transcription factor binding or histone modification or is directly related to gene expression (Banovich et al. 2014). Significantly, the overlap between mQTLs in brain tissue and disease SNPs was shown in schizophrenia (Hannon et al. 2015), and blood mQTLs were enriched in SNPs related to Crohn’s disease, hypertension, and rheumatoid arthritis (Gaunt et al. 2016). The overlap between mQTLs and disease SNPs with height, ulcerative colitis, Crohn’s disease, and coronary heart disease was also demonstrated in a recent report; however, substantial genetic variations in DNA methylation remain to be identified (McRae et al. 2018). Future studies may have to address issues related to low frequency and rare variants (Richardson et al. 2016).
Our study has several limitations, which are worth mentioning here. First, since the data were obtained only from cord blood, age-related changes could not be analyzed. Other studies investigated the effect of methylation over time and the effects of genetic variation and environmental changes with age. In fact, according to the studies conducted at five different stages in life, DNA methylation was affected by genetic and environmental factors; however, genetic effects decreased and environmental effects increased with time. To understand the impact of environment, we need chronological data, because DNA methylation changed over time (Moon et al. 2019). Second, our sample size is relatively small, and thus, it is difficult to generalize our results. Mapping genetic effects requires a much larger sample size. The lack of direct data supporting the relationship with disease is another limitation of our study. Therefore, in addition to the analysis of the mQTL, a future study of the distribution of mQTL according to the disease is desirable.
Conclusion
This study identified cis- and trans-mQTLs from cord blood obtained from a Korean birth cohort. We found a number of significant correlations between genotypes and methylations around functional genes such as ZFP57, TBX6, and PRDM8. These findings may provide some biological clues about pathogenesis mechanisms underlying several complex diseases via DNA methylations. It should be noted that our study provides the first mQTL results from Korean cord bloods.
References
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369
Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, McKenney SL, Witter F, Walston J, Feinberg AP, Fallin MD (2016) DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 11:354–362
Banovich NE, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y (2014) Methylation qtls are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 10:e1004663
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK (2011) DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 12:R10
Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, Kahn RS, Ophoff RA (2009) The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 4:e6767
Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, Buyske S, Schumacher FR, Peters U, Franceschini N, Ritchie MD et al (2013) Generalization and dilution of association results from European GWAS in populations of Non-European ancestry: the PAGE study. PLoS Biol 11:e1001661
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7
Chen W, Liu J, Yuan D, Zuo Y, Liu Z, Liu S, Zhu Q, Qiu G, Huang S, Giampietro PF (2016) Progress and perspective of TBX6 gene in congenital vertebral malformations. Oncotarget 7:57430
Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57
Consortium TMs (1999) Complete sequence and gene map of a human major histocompatibility complex. Nature 401:921
Delaneau O, Marchini J, Zagury J-F (2012) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179
den Dekker HT, Burrows K, Felix JF, Salas LA, Nedeljkovic I, Yao J, Rifas-Shiman SL, Ruiz-Arenas C, Amin N, Bustamante M et al (2019) Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J 53:1801795
Feinberg AP (2018) The key role of epigenetics in human disease prevention and mitigation. New Engl J Med 378:1323–1334
Ferguson-Smith AC (2011) Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 12:565
Gaunt TR, Shihab HA, Hemani G, Min JL, Woodward G, Lyttleton O, Zheng J, Duggirala A, McArdle WL, Ho K et al (2016) Systematic identification of genetic influences on methylation across the human life course. Genome Biol 17:61
Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK et al (2012) Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res 22:1395–1406
Gu Z, Gu L, Eils R, Schlesner M, Brors B (2014) circlize implements and enhances circular visualization in R. Bioinformatics 30:2811–2812
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, Troakes C, Turecki G, O’Donovan MC, Schalkwyk LC et al (2015) Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci 19:48
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13:86
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529
Howie B, Marchini J, Stephens M (2011) Genotype imputation with thousands of genomes. G3 Genes Genomes Genet 1:457–470
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955
Huang DW, Sherman BT, Lempicki RA (2008) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols 4:44
Johnson WE, Li C, Rabinovic A (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8:118–127
Joubert BR, Felix JF, Yousefi P, Bakulski KM, Just AC, Breton C, Reese SE, Markunas CA, Richmond RC, Xu C-J et al (2016) DNA methylation in newborns and maternal smoking in pregnancy: genome-wide Consortium Meta-analysis. Am J Hum Genet 98:680–696
Koh EJ, Hwang SY (2019) Multi-omics approaches for understanding environmental exposure and human health. Mol Cell Toxicol 15:1–7
Küpers LK, Monnereau C, Sharp GC, Yousefi P, Salas LA, Ghantous A, Page CM, Reese SE, Wilcox AJ, Czamara D et al (2019) Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat commun 10:1893
Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP (2008) Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 40:949
McRae AF, Powell JE, Henders AK, Bowdler L, Hemani G, Shah S, Painter JN, Martin NG, Visscher PM, Montgomery GW (2014) Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol 15:R73
McRae AF, Marioni RE, Shah S, Yang J, Powell JE, Harris SE, Gibson J, Henders AK, Bowdler L, Painter JN et al (2018) Identification of 55,000 replicated DNA methylation QTL. Sci Rep 8:17605
Min JL, Hemani G, Davey Smith G, Relton C, Suderman M (2018) Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics 34:3983–3989
Moon DH, Kwon SO, Kim WJ, Hong Y (2019) Identification of serial DNA methylation changes in the blood samples of patients with lung cancer. Tuberc Respir Dis 82:126–132
Richardson TG, Shihab HA, Hemani G, Zheng J, Hannon E, Mill J, Carnero-Montoro E, Bell JT, Lyttleton O, McArdle WL et al (2016) Collapsed methylation quantitative trait loci analysis for low frequency and rare variants. Hum Mol Genet 25:4339–4349
Shabalin AA (2012) Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28:1353–1358
Shi J, Marconett CN, Duan J, Hyland PL, Li P, Wang Z, Wheeler W, Zhou B, Campan M, Lee DS et al (2014) Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nat commun 5:3365
Stricker SH, Köferle A, Beck S (2016) From profiles to function in epigenomics. Nat Rev Genet 18:51
Takahashi N, Coluccio A, Thorball CW, Planet E, Shi H, Offner S, Turelli P, Imbeault M, Ferguson-Smith AC, Trono D (2019) ZNF445 is a primary regulator of genomic imprinting. Genes Dev 33:49–54
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S (2012) A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29:189–196
Turnbull J, Girard J-M, Lohi H, Chan EM, Wang P, Tiberia E, Omer S, Ahmed M, Bennett C, Chakrabarty A (2012) Early-onset Lafora body disease. Brain 135:2684–2698
Turner SD (2018) qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J Open Source Softw 3:731. https://doi.org/10.21105/joss.00731
Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. New Engl J Med 372:341–350
Xu Z, Niu L, Li L, Taylor JA (2015) ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res 44:e20–e20
Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82
Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44:369
Funding
This study was supported by the Korean Environment Industry & Technology Institute (KEITI) through “the Environmental Health Action Program”, funded by Korea Ministry of Environment (2017001360005).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Statement of human and animal rights
This study received ethical approval of the Kangwon National University Hospital IRB (B-2017-11-006).
Informed consent
Written informed consent was provided by each participant.
Conflict of interest
All authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Park, J., Kwon, S.O., Kim, SH. et al. Methylation quantitative trait loci analysis in Korean exposome study. Mol. Cell. Toxicol. 16, 175–183 (2020). https://doi.org/10.1007/s13273-019-00068-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-019-00068-3