
Abstract
We report a case of 2-year-old female with lateral ventricular glioma harboring both H3F3A K27M and BRAF V600E mutations. By the methylation analysis, the tumor was classified as a diffuse midline glioma H3 K27M mutant, WHO grade IV. However, the tumor was pathologically low-grade and likely localized rather than diffusely infiltrating. Further, the patient has survived more than 8 years after gross total resection of the tumor. Whereas both H3F3A K27M and BRAF V600E have been reported as poor prognostic markers in pediatric glioma, our case, along with several other reported cases, suggests that the coexistence of these two mutations might not indicate poor prognosis. The case emphasizes the importance of comprehensive assessment based on pathological, genetic and clinical findings and calls for further investigations of non-diffuse glioma with H3F3A K27M and glioma with H3F3A K27M and BRAF V600E.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The importance of genetic analysis of pediatric brain tumors has dramatically increased in recent years. First, diagnosis based on the revised 2016 WHO classification of tumors of the central nervous system requires the analysis of certain missense mutations, fusion genes, and amplifications/deletions of certain chromosome regions [1, 2]. Second, for medulloblastomas, a molecular classification is used both as a prognostic marker and for stratification into risk groups [3]. Third, some genetic abnormalities, such as the BRAF V600E mutation, and the NTRK, MET, and ALK/ROS1 fusion genes, can be effective therapeutic targets [4,5,6,7]. In addition, methylation-based classification has become a powerful tool [8]. For example, posterior fossa ependymoma is molecularly classified into the PFA and PFB subgroups, which have distinct clinical features [9,10,11]. Strum et al. reported that CNS–PNET could be reclassified into several novel subgroups [12]. Further, in 2018, the German Cancer Research Center (DKFZ) demonstrated the utility and accuracy of methylation-based classification of all brain tumor entities, and this classification tool is now available on their website [13].
Here, we report a case of pediatric midline low-grade glioma (LGG) in the lateral ventricle harboring both the H3F3A K27M and BRAF V600E mutations. Based on the methylation analysis, the tumor was classified as a “diffuse midline glioma with an H3 K27M mutant.” However, the pathological and clinical features were distinct from those of this entity.
Clinical summary
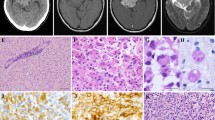
A 2-year-old female was referred to our hospital because of an intraventricular mass. The patient was born at 33 weeks gestation and she stayed in the hospital for 1 month because of her low birth weight. At the age of 1 year, she presented with epilepsy, which was controlled with an antiepileptic drug. MRI showed a nodular mass in the left ventricle (Fig. 1a–d). The patient underwent gross total resection of the mass because it was slowly growing. Based on the intraoperative findings, the border between the tumor and the normal thalamic tissues was partially unclear, whereas that between the tumor and ventricular lumen was clear. The local pathological diagnosis based on the 2007 WHO classification was oligoastrocytoma. She received no chemotherapy or radiotherapy and has survived without the evidence of disease for more than 8.5 years. After the publication of the revised fourth edition of the WHO classification in 2016, a molecular analysis and a review of the central pathology were performed (Fig. 2, 3a–f).

Radiological imaging of the case. a–d T2-weighted (a, b) and contrast-enhanced T1-weighted MRI (c, d) demonstrating a circumscribed, partially enhanced mass in the left ventricle

Pyrosequencing results showing the BRAF V600E and H3F3A K27M mutations
Pathological findings
The tumor consisted of moderately cellular glial proliferation within a fibrillary background. The lesional cells were relatively uniform in appearance and were round, oval, and spindle shaped. Pleomorphic, giant, epithelioid, rhabdoid, and piloid cells were lacking. The round cell component was associated with a perinuclear halo and resembled oligodendroglial tumors, whereas the spindle cells resembled the astrocytic lineage (Fig. 3a, b). There were a small number of rosette-like glial structures (Fig. 3c). Dysmorphic ganglion cells were absent. Mitotic figures were not detected. Necrosis, microvascular proliferation, Rosenthal fibers, and eosinophilic granular bodies were not observed. Focal microcalcification was present. Immunohistochemically, the tumor cells were positive for glial fibrillary acidic protein and S100 protein but were negative for epithelial membrane antigen and synaptophysin. In agreement with the molecular findings, they were immunopositive for H3 K27M, with a concomitant loss of H3K27me3 expression (Fig. 3d, e). BRAF V600E immunohistochemistry was positive (Fig. 3f), and H3 K27M and BRAF V600E staining appeared to be present in the same cells. The tumor retained ATRX expression, and p53 staining showed wild-type labeling. The MIB1 labeling index was 6.8%. The tumor mostly lacked transgressing neurofilament–positive fibers, and there was no evidence of diffuse infiltration.

Histological and immunohistochemical findings. The tumor consisted of glial proliferation with oligodendroglial (a) and astrocytic (b) morphology. There were a small number of rosette-like glial structures (c). Immunohistochemically, the tumor cells were positive for H3 K27M (d), with a concomitant loss of H3K27me3 expression (e). BRAF (V600E) immunohistochemistry was also diffusely positive (f)
Molecular analysis
DNA and RNA were extracted from frozen tumor tissue using the DNeasy Blood and Tissue Kit (Qiagen, Tokyo, Japan) and the miRNeasy Mini Kit (Qiagen, Tokyo, Japan), respectively. Hot spot mutations, including IDH1 R132, IDH2 R172, BRAF T599, BRAF V600, H3F3A K27, H3F3A G34, TERT promoter C250, TERT promoter C228, FGFR1 N546, and FGFR1 K656, were analyzed by pyrosequencing using the AQ assay with a PyroMark Q96 (Qiagen, Tokyo, Japan). The polymerase chain reaction (PCR) for pyrosequencing and the pyrosequencing assay were performed as previously described [14]. Primer sequences are listed in supplementary Table 1. The BRAF V600E and H3F3A K27M mutations were detected at frequencies of 40% and 46%, respectively (Fig. 2). The hot spot mutation in other analyzed genes was not detected. Multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA probemix P088-C2 (MRC-Holland, Amsterdam, the Netherlands) revealed no CDKN2A deletion. The existence of KIAA1549-BRAF fusion was assessed by reverse transcriptase PCR using previously reported primers and it was not detected [15].
A methylation classifier assay was then performed. DNA methylation was analyzed using an Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA), and the IDAT-files from the sample were uploaded to the online classifier developed by the DKFZ (https://www.molecularneuropathology.org/mnp). The tumor was classified as a “diffuse midline glioma, H3 K27M mutant” with a calibrated score of 0.98.
Discussion
Here, we report a case of midline glioma harboring concurrent H3F3A K27M and BRAF V600E mutations. The tumor was classified as a “diffuse midline glioma, H3 K27M mutant, WHO grade IV” by methylation analysis. However, in several aspects, the case was not entirely compatible with typical diffuse midline gliomas with H3F3A K27M mutations. Importantly, the present tumor was likely localized rather than diffusely infiltrating. Radiologically, it seemed well circumscribed; that is most of the tumor surface being surrounded by cerebral fluid. However, the border between tumor and normal thalamic tissue was partly unclear. Histologically, there was a paucity of transgressing neurofilament, suggesting a non-diffuse process, although this finding needs to be interpreted with caution because it may simply reflect the intra-ventricular growth. “Diffuse midline glioma with H3 K27M mutant WHO grade IV” was introduced as a new entity of WHO classification 2016, based on the knowledge at that time that recurrent mutation at K27 in H3F3A, HIST1H3B, and HIST1H3C is detected in high-grade glioma (HGG) from the pons, thalamus and spinal cord and that these mutations occur exclusively in diffuse midline gliomas, as stated in WHO blue book [2, 16]. However, some cases of non-diffuse glioma with H3F3A K27M were subsequently reported [17, 18]. Accordingly, Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy—Not Official WHO (cIMPACT-NOW) up date 2 emphasizes that “diffuse,” “midline,” “glioma” and “H3 K27M-mutant” are requirements for the diagnosis of diffuse midline glioma with H3 K27M [16]. Therefore, based on the cIMPACT-NOW-modified view of the WHO Classification, the present case seems incompatible with the “diffuse mid-line glioma with H3 K27M” designation. Whereas some of the reported localized gliomas with H3 K27M demonstrated the histology of well-established entities, the present case was difficult to classify histologically. The original diagnosis of oligoastrocytoma is likely invalid due to its mostly localized nature. Although histological findings, along with the presence of the BRAF mutation, may suggest a possibility of pilocytic astrocytoma, characteristic features of that entity were mostly lacking, including piloid or microcystic loose tissues, Rosenthal fibers, eosinophilic granular bodies, and microvascular proliferation. The morphology did not fit well with other BRAF-mutant localized gliomas either, such as gangliogioma and pleomorphic xanthoastrocytoma. Taken together, the present case would be best labeled descriptively as low-grade glioma, likely localized, not elsewhere classified as per the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy—Not Official WHO cIMPACT-NOW update 1 [16, 19].
Classification based on methylation profiles is a highly robust method for facilitating diagnosis and subgrouping brain tumors. However, the interpretation of methylation-based classification may sometimes require caution. H3 K27M would cause a considerable change in the tumor’s epigenetic landscape, which may mask fine differences among K27M-harboring tumors, some of which are not compatible with the definition of diffuse midline glioma with H3 K27M mutation, as in our case. Other mutations, such as BRAF, may on the other hand less affect epigenetic landscape, which could make them more difficult to distinguish by methylation profiling alone. The principle of defining a tumor entity, whether using should histology, genetics, methylation, or their combination, warrants further discussion.
In addition to the location and histological findings presented above, the patient’s age at diagnosis and her relatively long survival of the present case are also unusual for H3F3A K27M–positive glioma. The H3F3A K27M mutation is characteristically detected in pediatric gliomas. The mean age at diagnosis is 6–10 years [6, 20,21,22]. Most H3F3A K27M -positive gliomas arise in the pons or thalamus, and only approximately 5% arise in the ventricle [6, 20]. In most cases, the histological grade is high. The median overall survival of H3F3A K27M-positive midline glioma is less than 1 year, which is significantly worse than that of H3F3A K27 wild-type glioma. Based on an analysis of 77 pediatric patients with diffuse midline glioma over the age of 3 years, Karremman et al. reported that H3 K27M status has a negative impact on survival, regardless of tumor location or pathological grade [20]. There is a case report of pediatric LGG in the thalamus harboring H3 K27M transformed to HGG [23]. Pratt et al. also reported that H3 K27M has a negative impact on survival in circumscribed/non-diffuse glioma [24].
In contrast to H3F3A K27M, BRAF V600E is detected in a variety of tumors arising in patients of all ages, including thyroid and colorectal cancers as well as Langerhans cell histiocytosis [3, 25,26,27,28,29,30,31]. It is also detected in various glioma subtypes arising in any location, and the frequency is high in some subtypes, such as pleomorphic xanthoastrocytoma. The prognostic implications of BRAF V600E are dependent on various factors, including patient age, patient gender, tumor type, and the presence of other concurrent molecular abnormalities [3, 25,26,27,28,29,30,31,32]. For pediatric LGG, several reports suggest that BRAF V600E is associated with an aggressive clinical course, although this is somewhat controversial [3, 28, 32, 33]. Lassaletta et al. reported that the progression-free survival of patients with BRAF V600E–positive LGG is significantly worse than that of patients with wild-type BRAF LGG [3]. Mistry et al. reported that the frequency of BRAF V600E in pediatric LGG that underwent malignant transformation was higher than the frequency in non-transformed LGG (44% vs. 6%) [31].
Although the coexistence of H3F3A K27M and BRAF V600E mutations is rare, there have been several reported cases of pediatric glioma harboring these concurrent mutations (Table 1) [20, 22, 32, 34,35,36,37,38]. Interestingly, these include both diffuse midline glioma and non-diffuse glioma. In view of the diagnostic and prognostic information of this alternation, the recently published cIMPACT-NOW update 4 proposed the new classification name “Diffuse glioma, BRAF V600E mutant.” The diagnosis of some cases shown in Table 1 may warrant further discussion [39]. It is also noteworthy that the prognosis of these patients was not necessarily dismal but instead appeared to be somewhat better than that of patients with midline glioma harboring only the H3F3A K27M mutation, and that no patients died within the first year after the initial diagnosis. Notably, four patients, including all three patients under the age of 3 years, survived for more than 5 years. However, the number of reported cases with concurrent mutations is small and histological diagnosis, grading and age of diagnosis are variable. The follow-up periods are too short and recent widespread application of BRAF-targeted therapy may change their survival [40]. Therefore, further analysis of more cases is required to clarify these points.
In summary, we report a case of a relatively long-term survivor of pediatric midline glioma harboring concurrent BRAF V600E and H3F3A K27M. Although the tumor was classified as “diffuse midline glioma with H3K27M mt, WHO grade IV” based on methylation analysis, the pathological and clinical features of this case did not fit those of “diffuse midline glioma with H3 K27M mt” as defined in the current WHO classification (2016). Our case points to certain limitation of the current methylation classifier and it calls for further discussion of the integrated diagnosis of brain tumor. Further investigations of non-diffuse glioma with H3F3A K27M and glioma with both BRAF V600E and H3F3A K27 M are warranted.
References
Komori T (2017) Updated 2016 WHO classification of tumors of the CNS: turning the corner where molecule meets pathology. Brain Tumor Pathol 34(4):139–140
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016) WHO classification of tumours of the central nervous system, 2nd edn. IARC Press, Lyon
Ramaswamy V, Taylor MD (2017) Medulloblastoma: from myth to molecular. J Clin Oncol 35(21):2355–2363
Lassaletta A, Zapotocky M, Mistry M et al (2017) Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol 35(25):2934–2941
Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, Verhaak RG (2015) The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34(37):4845–4854
Nakano Y, Tomiyama A, Kohno T et al (2018) Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 36(1):14–19
Wu G, Diaz AK, Paugh BS et al (2014) The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46(5):444–450
Mack SC, Northcott PA (2017) Genomic analysis of childhood brain tumors: methods for genome-wide discovery and precision medicine become mainstream. J Clin Oncol 35(21):2346–2354
Witt H, Mack SC, Ryzhova M et al (2011) Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20(2):143–157
Mack SC, Witt H, Piro RM et al (2014) Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506(7489):445–450
Fukuoka K, Kanemura Y, Shofuda T et al (2018) Significance of molecular classification of ependymomas: c11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6(1):134
Sturm D, Orr BA, Toprak UH et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164(5):1060–1072
Capper D, Jones DTW, Sill M et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555(7697):469–474
Arita H, Narita Y, Matsushita Y et al (2015) Development of a robust and sensitive pyrosequencing assay for the detection of IDH1/2 mutations in gliomas. Brain Tumor Pathol 32(1):22–30
Jones DT, Kocialkowski S, Liu L et al (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68(21):8673–8677
Louis DN, Giannini C, Capper D et al (2018) cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol 135(4):639–642
Kleinschmidt-DeMasters BK, Mulcahy Levy JM (2018) H3 K27 M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin Neuropathol 37:53–63
Morita S, Nitta M, Muragaki Y et al (2018) Brainstem pilocytic astrocytoma with H3 K27M mutation: case report. J Neurosurg 129(3):593–597
Louis DN, Wesseling P, Paulus W et al (2018) cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol 135(3):481–484
Karremann M, Gielen GH, Hoffmann M et al (2018) Diffuse high-grade gliomas with H3 K27M mutations carry a dismal prognosis independent of tumor location. Neuro Oncol 20(1):123–131
Khuong-Quang DA, Buczkowicz P, Rakopoulos P et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124(3):439–447
Mackay A, Burford A, Molinari V et al (2018) Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell 33(5):829–842 (e825)
Ishibashi K, Inoue T, Fukushima H, Watanabe Y et al (2016) Pediatric thalamic glioma with H3F3A K27M mutation, which was detected before and after malignant transformation: a case report. Childs Nerv Syst 32(12):2433–2438
Pratt D, Natarajan SK, Banda A et al (2018) Circumscribed/non-diffuse histology confers a better prognosis in H3K27M-mutant gliomas. Acta Neuropathol 135(2):299–301
Tabouret E, Bequet C, Denicolai E et al (2015) BRAF mutation and anaplasia may be predictive factors of progression-free survival in adult pleomorphic xanthoastrocytoma. Eur J Surg Oncol 41(12):1685–1690
Vuong HG, Altibi AMA, Duong UNP et al (2018) BRAF mutation is associated with an improved survival in glioma-a systematic review and meta-analysis. Mol Neurobiol 55(5):3718–3724
Chen X, Pan C, Zhang P et al (2017) BRAF V600E mutation is a significant prognosticator of the tumour regrowth rate in brainstem gangliogliomas. J Clin Neurosci 46:50–57
Jones DTW, Witt O, Pfister SM (2018) BRAF V600E status alone is not sufficient as a prognostic biomarker in pediatric low-grade glioma. J Clin Oncol 36(1):96
Kim TH, Park YJ, Lim JA et al (2012) The association of the BRAF (V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer 118(7):1764–1773
Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, Pai S, Bishop J (2014) BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol 32(25):2718–2726
Heritier S, Emile JF, Barkaoui MA et al (2016) BRAF mutation correlates with high-risk Langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol 34(25):3023–3030
Mistry M, Zhukova N, Merico D et al (2015) BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33(9):1015–1022
Ho CY, Mobley BC, Gordish-Dressman H et al (2015) A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130(4):575–585
Zhang J, Wu G, Miller CP et al (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45(6):602–612
Pages M, Beccaria K, Boddaert N et al (2018) Co-occurrence of histone H3 K27M and BRAF V600E mutations in paediatric midline grade I ganglioglioma. Brain Pathol 28(1):103–111
Nguyen AT, Colin C, Nanni-Metellus I et al (2015) Evidence for BRAF V600E and H3F3A K27M double mutations in paediatric glial and glioneuronal tumours. Neuropathol Appl Neurobiol 41(3):403–408
Solomon DA, Wood MD, Tihan T, Bollen AW, Gupta N, Phillips JJ, Perry A (2016) Diffuse midline gliomas with histone H3-K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 26(5):569–580
Ryall S, Krishnatry R, Arnoldo A et al (2016) Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun 4(1):93
Ellison DW, Hawkins C, Jones DTW et al (2019) cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol 137(4):683–687
Penman CL, Faulkner C, Lowis SP, Kurian KM (2015) Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front Oncol 5:54
Acknowledgements
The authors thank the patient and her family for participating in this research. We also thank Y. Matsushita for technical support with the molecular analysis and K. Fukuoka for insightful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest associated with this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nakano, Y., Yamasaki, K., Sakamoto, H. et al. A long-term survivor of pediatric midline glioma with H3F3A K27M and BRAF V600E double mutations. Brain Tumor Pathol 36, 162–168 (2019). https://doi.org/10.1007/s10014-019-00347-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-019-00347-w