
Abstract
The proto-oncogene tyrosine-protein kinase ROS1 (ROS1) is a tyrosine kinase that is closely related to anaplastic lymphoma kinase receptor (ALK). We describe a novel KLC1–ROS1 fusion identified in a case of pediatric low-grade glioma. This was detected by RNA sequencing and confirmed by reverse-transcription PCR and fluorescent in situ hybridization. Immunohistochemical staining for ROS1 was positive in the tumor cytoplasm. In vitro analysis demonstrated the oncogenic activity of this fusion, which was suppressed by the ALK/ROS1 inhibitor, crizotinib. Our case and others suggest that various ROS1 fusions might be present in a subset of pediatric gliomas, which could be targeted for therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
ROS1, located on chromosome 6, encodes the proto-oncogene ROS1 [1, 2]. As an orphan receptor tyrosine kinase, it is closely related to anaplastic lymphoma kinase (ALK). Both have an extracellular domain, a transmembrane region, and an intracellular tyrosine kinase domain, and are involved in the activation of the JAK/STAT, RAS/MAPK, and PI3K pathways. The first ROS1 fusion, GOPC-ROS1, was discovered in a human glioblastoma cell line (U118MG). Since then, more than 25 ROS1 fusions with various 5′ fusion partners have been detected in diverse types of cancers including non-small lung cancer and cholangiocarcinomas; however, they are not often found in clinical samples of adult glioblastoma [1, 3]. ROS1 fusion genes invariably retain the kinase domain of ROS1. The important clinical implication of ROS1 fusions is that they can be targeted by ROS1 inhibitors [2]. Here, we present a novel KLC1–ROS1 fusion identified in a case of pediatric low-grade glioma. In vitro analysis demonstrated that the fusion has oncogenic activity that can be suppressed by an ROS1 inhibitor.
Clinical summary
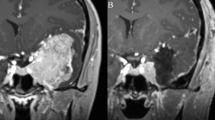
A 3-year-old girl was suspected of having a brain tumor when she received a computed tomography scan after a minor head injury (Fig. 1a). The patient was neurologically asymptomatic. Magnetic resonance imaging showed a slightly gadolinium-enhanced mass (5.7 × 4.0 × 3.8 cm) located in the right frontal lobe (Fig. 1b, c). The patient underwent a gross total tumor resection and experienced no recurrence for 7 years.

Clinical and histopathological features of pediatric glioma patient and detection of KLC1–ROS1 fusion. a–c Computed tomography (CT) and magnetic resonance imaging (MRI) demonstrated a partially enhanced mass in the right frontal lobe. Histologically, the tumor showed vague internal multinodularity (d) and was composed of cellular astrocytic proliferation embedded within a dense compact background of glial filaments. Most tumor cells were gemistocyte in appearance (e), whereas a minor subset of cells was of fibrillary and spindle-type (f). Hematoxylin and eosin stain. Magnifications and scale bars are ×100, 200 µm, ×600, 50 µm, and ×200, 50 µm respectively (for d–f). gKLC1–ROS1 fusion was validated by Sanger sequencing of the PCR product. h Immunohistochemical analysis showed diffuse cytoplasmic ROS1 protein expression (×400 magnification; scale bar = 50 µm). i Fluorescent in situ hybridization analyses using ROS1 break-apart probes showed an isolated green (3′) signal in addition to the intact red/green fused signal, indicating an ROS1 rearrangement (left). Two red/green fused signals per nucleus were detected in the fusion-negative control (right)
Pathological findings
The tumor was sharply demarcated from the surrounding brain tissue. It showed vague internal multinodularity and was composed of cellular astrocytic proliferation embedded within a dense compact background of glial filaments (Fig. 1d). Most tumor cells were gemistocyte in appearance and harbored relatively uniform, vesicular, non-hyperchromatic nuclei (Fig. 1e). A minor subset of cells was fibrillary and spindle type (Fig. 1f). Mitotic figures were not observed. No dysmorphic ganglion cells were present, nor was there morphological evidence of ependymal differentiation. No necrosis or microvascular proliferation was present. Rosenthal fibers and eosinophilic granular bodies were not observed. There was a focal area where numerous minute calcospherites were deposited. Immunohistochemically, the tumor cells were positive for glial fibrillary acidic protein, S100 protein, synaptophysin (focal, dot-like), Olig2 (weak, focal), epithelial membrane antigen (focal, dot-like), p53 (focal, wild-type pattern), and ATRX (retained), whereas they were negative for CD34. No CD34-positive ramifying elements were observed. The MIB-1 labeling index was approximately 4.3%. The non-infiltrative nature of the tumor was highlighted by the lack of transgressing neurofilament-positive fibers within the tumor tissue.
Molecular analysis
Identification of KLC1–ROS1 fusion
Targeted sequencing of all coding exons of 93 selected brain tumor-related genes using DNA isolated from the tumor sample revealed no significant somatic pathogenic variants (Supplementary Information S1 and Supplementary Table S1). We then performed RNA sequencing (Supplementary Information S1) and a KLC1–ROS1 fusion was detected. The presence of this fusion was confirmed by reverse-transcription PCR followed by Sanger sequencing (Fig. 1g; Supplementary Information S1). Immunohistochemistry (IHC) for ROS1 was diffusely positive in the tumor cytoplasm (Fig. 1h). Fluorescence in situ hybridization (FISH) using a ROS1 break-apart probe set showed the presence of an ROS1 rearrangement with one fused (5′/3′) signal and isolated 3′ green signals (Fig. 1i; Supplementary Information S1).
Oncogenic activity of KLC1–ROS1 fusion
To investigate the oncogenic activity of the KLC1–ROS1 fusion in glioma cells, we introduced the fusion into NIH/3T3 fibroblasts by lentiviral transduction and performed focus forming assays. Cells expressing the fusion demonstrated remarkable focus forming ability compared to control cells (Fig. 2a). Subsequently, to examine fusion-mediated oncogenic molecular signaling and phenotypes in glioblastoma cells, we generated two fusion mutants including an ROS1 kinase-dead mutant (KD) and a deletion mutant of the KLC1 portion (KLC1-del) of fusion (Fig. 2b). Fusion and mutant constructs were transiently introduced into A172 human glioblastoma cells, and lysates were analyzed by immunoblotting. The fusion, but not the ROS1 kinase-dead (KD) and KLC1-deletion (KLC1-del) mutants, induced prominent activation of JAK-STAT signaling, which is a downstream oncogenic pathway activated by ROS1, as shown by elevated JAK2 and STAT3 phosphorylation (Fig. 2c). In addition, A172 cells transiently expressing the fusion, but not the empty vector and the two mutants, exhibited increased cell proliferation and migration, which were suppressed by crizotinib, a clinically available ROS1 inhibitor (Fig. 2d). These results suggested that the KLC1–ROS1 fusion is a transforming oncogene, and that both the KLC1 portion and ROS1 kinase activities are essential for its oncogenic activity.

Functional analysis of the KLC1–ROS1 fusion. a NIH3T3 cells were infected with a lentiviral vector expressing the empty vector (control) or the wild-type KLC1–ROS1 fusion, as indicated, and analyzed by focus forming assays. The magnified images of each group of treated cells obtained by phase-contrast microscopy are shown. b Schematic illustration of KCL1-ROS1 mutants generated in this study. The number indicates amino acid (aa) position and the breakpoints are indicated by arrows. c Indicated KLC1–ROS1 constructs were expressed in A172 glioma cells. After 72 h, the cell lysates were analyzed by immunoblotting using indicated antibodies. A GAPDH antibody was used to control for protein loading. d (Upper panel) indicated KLC1–ROS1 constructs were expressed in A172 glioma cells after treatment with vehicle (DMSO) or the ROS1 inhibitor crizotinib (500 nM). After 72 h, the number of cells in each treatment group was analyzed by proliferation assays. *p < 0.01. (Lower panel) indicated that KLC1–ROS1 constructs were expressed in A712 glioma cells. After 72 h, the cells were analyzed by migration assays in the presence of vehicle (DMSO) or crizotinib (500 nM). *p < 0.01
Discussion
A novel KLC1–ROS1 fusion was identified in a case of pediatric low-grade localized glioma; in vitro analysis demonstrated its oncogenic activity. No other pathogenic variants were detected based on the analysis of 93 genes associated with brain tumors. Hence, this novel fusion was likely a primary driver mutation in this case.
Kinesin light chain 1 (KLC1), encoded by the KLC1 gene on chromosome 14, is involved in intracellular transport systems [4]. KLC1-ALK fusions have recurrently been detected in lung cancer, and the breakpoint of KLC1 is the same as that found in the KLC1–ROS1 fusion identified in this report, specifically, the 3′ end of exon 9 of KLC1 [5, 6]. Furthermore, the coiled-coil domain of KLC1 was retained in these fusions. The mechanism through which each fusion partner contributes to continuous activation of the kinase domain of ROS1 is not fully understood. However, more than half of previously reported ROS1 partners had a coiled-coil domain, and dimerization via these domains is thought to contribute to activation of the ROS1 kinase domain [2].
Interestingly, ALK or ROS1 rearrangements have been recently reported in a subset of pediatric brain tumors (Table 1) [7,8,9,10,11,12]. These patients tended to be infants and younger children, although the number of the reported cases was small. Patients with these rearrangements might benefit from crizotinib or next-generation ALK/ROS1 inhibitors, which have shown promising activities against lung tumors that metastasize to the CNS [12, 13]. These targeted therapies might serve as an ideal alternative to the conventional chemotherapy and radiotherapy, which unfavorably affect the growth and development of children.
RNA sequencing is currently the most comprehensive and ideal method to detect fusion genes. However, it is time- and cost-consuming, and might not be routinely available in the clinical setting. Therefore, it is necessary to establish a practical approach to screen gene fusions. ALK or ROS1 rearrangements can be detected by FISH. However, GOPC–ROS1 fusions, which have been recurrently observed in pediatric glioma, can be difficult to detect by FISH analysis, because both genes are closely located on 6q22.1 and a specifically designed probe set is required for its detection [2, 14]. These facts highlight the importance of probe design. In fusion-positive cases, ALK or ROS1 protein expression can be detected by IHC. We previously established this protocol using a large number of lung carcinomas whose ROS1 rearrangement status was available [15]. In addition, we previously used the same protocol to successfully identify ROS1-rearranged inflammatory myofibroblastic tumors [16, 17]. With regard to the CNS tumors, one study reported that ROS1 IHC using the same antibody clone showed no ROS1 reactivity in 109 glioblastomas that lacked ROS1 fusions by FISH [3]. We, too, have not seen ROS1 IHC reactivity in more than 50 glioblastomas that lacked ROS1 rearrangement by FISH (unpublished data). Taken together, it is eminently possible that ROS1 IHC could serve as a screening tool or surrogate for ROS1-rearranged tumors in the CNS. However, future studies, including a large number of CNS tumors of different types, are necessary to test its utility.
Thus, it is recommended that both FISH and IHC be performed, followed by RNA sequencing when applicable.
In summary, a novel KLC1–ROS1 fusion has been identified in a case of pediatric low-grade, localized glioma. Further investigations will help to determine any common features of brain tumors harboring ROS1 rearrangements and which patients should receive screening tests; moreover, the therapeutic benefit and appropriate use of ROS1 inhibitors for these patients should be validated in clinical trials.
References
Davies KD, Doebele RC (2013) Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res 19:4040–4045
Uguen A, De Braekeleer M (2016) ROS1 fusions in cancer: a review. Future Oncol 12:1911–1928
Lim SM, Choi J, Chang JH et al (2015) Lack of ROS1 gene rearrangement in glioblastoma multiforme. PLoS One 10:e0137678
Verhey KJ, Hammond JW (2009) Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol 10:765–777
Togashi Y, Soda M, Sakata S et al (2012) KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One 7:e31323
Wang P, Xiao P, Ye Y et al (2017) Rapid response of brain metastasis to crizotinib in a patient with KLC1-ALK fusion and MET gene amplification positive non-small cell lung cancer: a case report. Cancer Biol Med 14:183–186
Johnson A, Severson E, Gay L et al (2017) Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22:1478–1490
Coccé MC, Mardin BR, Bens S et al (2016) Identification of ZCCHC8 as fusion partner of ROS1 in a case of congenital glioblastoma multiforme with a t(6;12)(q21;q24.3). Genes Chromosomes Cancer 55:677–687
Olsen TK, Panagopoulos I, Meling TR et al (2015) Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro Oncol 17:1365–1373
Aghajan Y, Levy ML, Malicki DM et al (2016) Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep. https://doi.org/10.1136/bcr-2016-217189
Kiehna EN, Arnush MR, Tamrazi B et al (2017) Novel GOPC(FIG)-ROS1 fusion in a pediatric high-grade glioma survivor. J Neurosurg Pediatr 20:51–55
Rossing M, Yde CW, Sehested A et al (2017) Genomic diagnostics leading to the identification of a TFG-ROS1 fusion in a child with possible atypical meningioma. Cancer Genet 212–213:32–37
Cameron L, Solomon B (2015) New treatment options for ALK-rearranged non-small cell lung cancer. Curr Treat Options Oncol 16:49
Bubendorf L, Büttner R, Al-Dayel F et al (2016) Testing for ROS1 in non-small cell lung cancer: a review with recommendations. Virchows Arch 469:489–503
Yoshida A, Tsuta K, Wakai S et al (2014) Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol 27:711–720
Fujita H, Yoshida A, Taniguchi H et al (2015) Adult-onset inflammatory myofibroblastic tumour of the stomach with a TFG-ROS1 fusion. Histopathology 66:610–612
Yamamoto H, Yoshida A, Taguchi K et al (2016) ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 69:72–83
Acknowledgements
The authors thank the patient and their family for participating in this research. We thank T. Ushijima, S. Yamashita, and R. Nagano for their advice and technical support for DNA analysis. This work was supported in part by the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development, AMED [no. JP17ck0106168 (T.K. and K. I.)].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest associated with this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nakano, Y., Tomiyama, A., Kohno, T. et al. Identification of a novel KLC1–ROS1 fusion in a case of pediatric low-grade localized glioma. Brain Tumor Pathol 36, 14–19 (2019). https://doi.org/10.1007/s10014-018-0330-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10014-018-0330-3