
Abstract
The prokaryotic communities of four salterns (Bingöl, Fadlum, Kemah, and Tuzlagözü) in Turkey were examined and compared using the cultivation and cultivation-independent methods [fluorescence in situ hybridization (FISH) and 454 pyrosequencing]. FISH analysis with universal probes revealed that feeding waters carried 1.6 × 102–1.7 × 103 cells mL−1, while crystallization ponds carried 3.8 × 106–2.0 × 107 cells mL−1 that were mostly haloarchaea, including square cells (except for Kemah). High-throughput 16S rRNA-based gene sequencing showed that the most frequent archaeal OTUs in Bingöl, Fadlum, Tuzlagözü, and Kemah samples were affiliated with Haloquadratum (76.8 %), Haloarcula (27.8 %), Halorubrum (49.6 %), and Halonotius (59.8 %), respectively. Bacteroidetes was the dominant bacterial phylum in Bingöl and Fadlum, representing 71.5 and 79.5 % of the bacterial OTUs (respectively), while the most abundant bacterial phylum found in the Kemah saltern was Proteobacteria (79.6 %). The majority of the bacterial OTUs recovered from Tuzlagözü belonged to the Cyanobacteria (35.7 %), Bacteroidetes (35.0 %), and Proteobacteria (25.5 %) phyla. Cultivation studies revealed that the archaeal isolates were closely related to the genera Halobacterium, Haloarcula, and Halorubrum. Bacterial isolates were confined to two phyla, Proteobacteria (Alphaproteobacteria and Gammaproteobacteria classes) and Bacteroidetes. Comparative analysis showed that members of the Euryarchaeota, Bacteroidetes, Proteobacteria, and Cyanobacteria phyla were major inhabitants of the solar salterns.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hypersaline environments represented by hypersaline lakes, soils, springs, solar salterns, and rock salt deposits are widely distributed. Studies on hypersaline ecosystems have mainly focused on saline lakes and solar salterns (Benlloch et al. 2002; Casamayor et al. 2002; Burns et al. 2004; Maturrano et al. 2006; Mutlu et al. 2008; Baati et al. 2008; Makhdoumi-Kakhki et al. 2012). Solar salterns are considered model systems for ecological studies because of their low microbial diversity and relatively constant environmental conditions. Microbial communities of crystallizer ponds are well defined and characterized by dense populations of haloarchaeal species, particularly Haloquadratum (previously known as the square haloarchaeon of Walsby) and the extremely halophilic bacterium Salinibacter ruber, which is affiliated with the phylum Bacteroidetes (Antón et al. 2002; Burns et al. 2004; Oren and Rodrı́guez-Valera 2001; Oren 2008).
Erzincan and Sivas are provinces located east of Anatolia in Turkey. The Tertiary basin that extends between the two provinces is represented by deep sea, shallow sea, and lagunar and continental sediments, showing complex sedimentary and tectonic features, and began its formation in the Paleogene period (Aktimur et al. 1990; Çiner and Koşun 1996). Tuzlagözü, Fadlum, Bingöl (Sivas) and Kemah (Erzincan) salterns are found in this area, and consist of many evaporation ponds fed by hypersaline spring water. There are many reports related to the microbiota of coastal salterns fed by sea water (Benlloch et al. 2002; Burns et al. 2004; Baati et al. 2008; Sabet et al. 2009), but few studies of inland salterns fed by hypersaline spring water (Maturrano et al. 2006). These four salterns have no connection with the sea, but their salt compositions resemble thalassohaline environments (Oren 2002), derived from evaporated seawater, and are rich in sodium ions.
Currently, high-throughput sequencing methods allow us to comprehensively analyze the structure of microbial communities from different environmental samples and have recently been used to investigate the microbial compositions of different hypersaline environments (Ghai et al. 2011; Fernández et al. 2014). In the present work, we used pyrosequencing to reveal the microbial community composition of crystallizers and also investigated the microbiota inhabiting both the crystallizers and feeding waters using the cultivation and FISH-based analyses. The results obtained from analyzing crystallizers of the Tuzlagözü, Fadlum, and Bingöl salterns demonstrated many shared features between them as well as with those obtained from previous studies, while the microbiota of the Kemah salterns displayed a distinct profile.
Materials and methods
Sampling and physico–chemical analysis
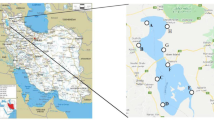
Water samples (5 L each) were collected from hypersaline spring waters and crystallization ponds associated with the Tuzlagözü (39°43″N, 37°40″E), Fadlum (39°41″N, 37°07″E), Bingöl (39°43″N, 37°07″E) (June, 2012), and Kemah salterns (39°39″N, 39°02″E) (August, 2011) (Fig. 1). Total salt concentrations of the samples were measured with a hand refractometer (Eclipse) and refractive indices were determined by a Bausch and Lomb refractometer. The pH of each sample was measured as well (Mettler Toledo, Schwerzenbach, Switzerland).

Gray circles on the map show sampling locations (Fadlum, Bingöl, Tuzlagözü, and Kemah salterns)
DAPI counts and FISH
Samples were treated with 7 % formaldehyde to fix cells, and then filtered (0.2 μm filters, GTTP, Millipore, MA, USA) to collect them. An optimized FISH protocol for extremely halophilic microorganisms was performed as described previously (Antón et al. 1999), using domain-specific probes; Arc915 for Archaea, and EUB338 for Bacteria (Amann et al. 1990, 1995). After hybridization and DAPI staining, the cells were counted using epifluorescence microscopy (Leica DM6000 M) and enumeration performed across approximately 30 microscopic fields per sample (Snaidr et al. 1997).
Nucleic acid extraction
100 milliliter of each sample was used for nucleic acid extraction using a modified version of the methods of Cifuentes et al. (2000) and Nogales et al. (1999), as previously described in detail (Mutlu et al. 2008). For the collection of microorganisms, water samples were filtered through a 0.22-µm pore size GV filter (Durapore, Millipore). Filters were then cut into pieces and mixed with extraction buffer (100 mM Tris–HCl, 100 mM EDTA pH 8.0), lysozyme (3 mg mL−1), and incubated at 37 °C. Proteinase K (150 mg mL−1) and 10 % sodium dodecyl sulfate were added and samples incubated at 37 °C. After the addition of 5-M NaCl and CTAB solution (10 % CTAB, 0.7 M NaCl), samples were treated with liquid nitrogen and then incubated in a water bath at 65 °C. Nucleic acid extraction and purification were performed using phenol–chloroform–isoamyl alcohol (25:24:1) followed by ethanol precipitation and resuspension in sterile Milli-Q water. Nucleic acid samples were, subsequently, stored at −80 °C.
Isolation and cultivation
Modified growth medium (MGM) (supplemented with 0.1 % yeast extract and 0.5 % peptone) with 12, 18, or 23 % total salt concentration was used to isolate microorganisms. Seawater medium (supplemented with 1 % yeast extract) with 25 % total salt concentration, DSMZ medium (containing 0.1 % yeast extract) for Salinibacter ruber, and DBCM2 medium (containing 0.1 % pyruvate) were also used (Dyall-Smith 2009). Water samples were directly used for inoculation as were serial 100-fold dilutions (10−2, 10−4, 10−6). Plates were incubated aerobically at 37 °C for 6 weeks. Colonies were picked and subsequently restreaked three times on the same medium. Isolates were then stored as 15 % glycerol stocks at −80 °C. Based on morphological features, some of the colonies were selected and suspended in sterile 100-μL Milli-Q water to extract DNA by boiling. For colonies, the DNA extraction protocol mentioned in the previous section was used. Polymerase chain reaction (PCR) amplification products of the 16S rRNA genes were obtained from each of the DNA extracts. Broad-range PCR primers specific for Bacteria and Archaea were used in these reactions; 21f (5′-TTCCGGTTGATCCTGCCGGA-3′) (DeLong 1992) specific for the Archaea domain and 27f (5′-AGAGTTTGATCATGGCTCAG-3′) (Lane et al. 1985) specific for the Bacteria domain were used as forward primers, while 1492r (5′-GGTTACCTTGTTACGACTT-3′) (Lane et al. 1985) was used as the reverse primer for both Bacteria and Archaea. Positive control DNA templates were also tested (Haloarcula hispanica for Archaea and Salinibacter ruber for Bacteria). PCR products were analyzed using agarose gel electrophoresis (1 % agarose gels) and depending on the presence of signal, isolates were classified as either Bacteria or Archaea. Amplified ribosomal DNA restriction analysis (ARDRA) was then used to further characterize the isolates. Briefly, the PCR products of the 16S rRNA genes were digested with the restriction endonucleases HinfI and MboI (Fermentas) at 37 °C overnight and digestion products visualized by agarose gel electrophoresis (2 % agarose gels). Resulting ARDRA patterns were analyzed and compared, and a representative from each distinct ARDRA pattern was selected for sequencing.
DNA sequence analysis and phylogenetic tree construction
PCR products of the 16S rRNA genes obtained from cultivated isolates were purified (Wizard® SV Gel and PCR Clean-Up System; Promega) and sequenced (CEQ™ 8000 DNA sequencer; Beckman Coulter, Fullerton, CA, USA). Sequences were compared with those present in the Ribosomal Database Project (RDP) (Cole et al. 2013) and screened for putative chimeras using Bellerophon (Huber et al. 2004). Sequences were aligned using MUSCLE (Edgar 2004), and phylogenetic tree reconstructions were performed using maximum likelihood and PhyML (bootstrap values calculated from 100 bootstrap replicates; Dereeper et al. 2008; Guindon et al. 2010).
Pyrosequencing and data analysis
Total DNA from the various water samples was sent to a commercial sequencing service (ChunLab Inc., Seoul, South Korea) for pyrosequencing targeting the V1–V3 region of the 16S rRNA gene using a Roche 454 GS FLX+/Junior system. Pyrosequencing reads were grouped into operational taxonomic units (OTUs) at 97 % sequence similarity using CD-HIT (Li and Godzik 2006). The EzTaxon-e database was then used for taxonomic classification (Kim et al. 2012, http://eztaxon-e.ezbiocloud.net/) and chimeric sequences removed with UCHIME (Edgar et al. 2011). Estimation of alpha diversity and community composition were performed with the CLcommunity™ program (ChunLab Inc., South Korea).
Nucleotide accession numbers
Sequences have been deposited in GenBank under accession numbers KF668248-66, KF976348-62, KF985241, and KJ787792-94.
Results
Sampling and physico–chemical analysis
The total salinity of the samples obtained from hypersaline spring waters and crystallization ponds was determined by a hand refractometer and ranged from 18 to 25 and 20 to 27 %, respectively. The refractive indices of the samples ranged between 1.3635 (accounting for ~20 g salt/100 g) and 1.3801 (accounting for ~26 g salt/100 g). pH was also measured and found to be approximately neutral (Table 1).
DAPI counts and FISH analysis
Total cell counts revealed that the number of microbial cells in the hypersaline spring waters was very low, in contrast to the crystallization ponds, in which total cells were above 106 cells mL−1 (Table 2). The Fadlum (27 % salinity), Bingöl (25 % salinity), and Tuzlagözü (20 % salinity) crystallization ponds were dominated by archaeal cells, comprising 94.2, 76.6, and 56.4 % of the community, respectively, based on an Archaea-specific probe. These results showed that with increased salinity, the archaeal population comprised a greater fraction of the community, except for the Kemah sample (27 % salinity), in which bacterial cells were the majority fraction (92.4 %), and the square cells that were observed within the Bingöl, Fadlum, and Tuzlagözü ponds were not present within the Kemah pond. Eukaryote-like cells were also observed, particularly in feeding waters (Fig. 2), but bacterial and archaeal cells comprised the majority of the communities (>84 %) in all four samples. In the Maras paper by Maturrano et al. (2006), there is also a figure showing eukaryotic-like cells in the spring water.

Images of the crystallizer microbiota (a–e). DAPI staining shown on the left, and hybridization signals with FISH probes Arc915 (a, b–d) and Eub338 (c–e) on the right. Cells in the water feeding the salterns (hypersaline spring waters) (f–g) show DAPI staining. Image sources were Bingöl (a), Tuzlagözü (b, c, f), Fadlum (d, g), and Kemah salterns (e)
Isolation of Bacteria and Archaea
Isolation of Bacteria and Archaea was performed using MGM and SW media across a range of salinity. Viable counts on MGM, with salt concentration at 18 and 23 %, ranged between 7.3 × 103 and 3.0 × 104 cfu mL−1 (crystallization ponds) and 2.4 × 102 and 1.5 × 103 cfu mL−1 (hypersaline spring waters). Viable counts were found to be affected by salt concentration in the medium. Media supplemented with 12 or 25 % salt yielded low numbers of colonies. No colonies appeared on plates with 25 % salt which were inoculated with hypersaline spring waters (except Kemah). Most of the bacterial isolates were moderately halophilic, growing optimally in the presence of 6–15 % salt. However, Salinibacter ruber and all archaeal strains were extremely halophilic and growth occurred optimally between 18 and 25 % salt. Halotolerant microorganisms were also isolated, such as Photobacterium strains. 393 isolates, consisting of 252 Bacteria and 141 Archaea, were selected for further study on the basis of their differing morphological features, salt responses and sampling locations. Analysis of their 16S rRNA genes by ARDRA revealed 9 different patterns within the archaeal isolates and 27 patterns for the bacterial isolates (supplementary Table S1).
The 16S rRNA genes of representative archaeal isolates from the nine ARDRA patterns were sequenced and found to be closely related to previously described haloarchaeal species. All fell within three genera: Haloarcula, Halorubrum, and Halobacterium. Representatives of these genera have been widely isolated from hypersaline environments (Benlloch et al. 2002; Burns et al. 2004; Maturrano et al. 2006; Sabet et al. 2009; Caton et al. 2009; Makhdoumi-Kakhki et al. 2012; Dillon et al. 2013). The most abundant haloarchaeon was Haloarcula isolated from the Fadlum, Tuzlagözü, and Bingöl salterns. Halobacterium salinarum was isolated from all of the crystallization ponds, except feeding waters, and was readily isolated on 25 % SW supplemented with 1 % yeast extract. Sequencing of bacterial representatives of the 27 ARDRA patterns revealed a wide diversity of genera, but all belonged to two phyla: Proteobacteria (Alphaproteobacteria and Gammaproteobacteria) and Bacteroidetes. Members of these phyla were also dominant in Chula Vista (USA) and Sfax, as previously reported (Baati et al. 2008; Zhaxybayeva et al. 2013). Salicola-related sequences showed high-sequence similarity with those isolated from Maras solar salterns fed by hypersaline spring water (Maturrano et al. 2006), isolated from both crystallization ponds and feeding waters, was the most frequent bacterial isolate (27.3 % of the bacterial isolates) in our study. Thalassospira- and Halomonas-related strains were also abundant. The remaining isolates were phylogenetically related to the genera Salinibacter, Rhodovibrio, Marinobacter, Idiomarina, Photobacterium, Vibrio, Chromohalobacter, Fodinibius, and Salinivibrio. 16S rRNA-encoding genes from some bacterial isolates had <95 % sequence similarity with previously cultured sequences, indicating that they may be representatives of novel genera obtained using modified growth medium with 12 % total salt concentration. The diversity of isolates at the genus-level is summarized in Fig. 3. Phylogenetic analysis was performed using PhyML. Sequences recovered from the isolates were aligned, and the phylogenetic trees for Archaea and Bacteria constructed based on complete or nearly complete 16S rRNA sequences using different algorithms are shown in Figs. 4 and 5.

Distribution of the isolates at genus level obtained from the crystallization ponds and hypersaline spring waters. No archaeal isolates were obtained from Kemah hypersaline spring water

Phylogenetic tree of members of the Bacteria was constructed by the maximum likelihood treeing algorithm. Black circles represent 16S rDNA sequences from isolates obtained. Sequence of Thermus filiformis strain DSM 4687 was used as an outgroup. Scale bar represents expected number of substitutions per site

Phylogenetic tree of members of the Archaea was constructed by maximum likelihood. Black circles represent 16S rDNA sequences from the isolates. Sequence of Methanococcus voltae strain A3 was used as an outgroup. Scale bar represents expected number of substitutions per site
Pyrosequencing, rarefaction analysis, and diversity indices
Pyrosequencing of all samples yielded a total of 40,995 high-quality reads with an average length of ~440 bp, and were subsequently clustered into operational taxonomic units (OTUs, 97 % similarity). Archaeal OTUs representing eight genera within the Class Halobacteria (Haloarcula, Halonotius, Halorubrum, Haloplanus, Halorhabdus, Natronomonas, Halomicroarcula, and Halorubellus) and bacterial OTUs representing seven genera within the Bacteroidetes, Proteobacteria, and Cyanobacteria phyla (Salinibacter, Aliifodinibius, Spiribacter, Acinetobacter, Halovibrio, Salicola, and Euhalothece) were shared between the four salterns (Fig. 6). The most abundant bacterial OTUs affiliated with Salisaeta (54.3 %), Salinibacter (68.9 %), Euhalothece (34.3 %), and Pseudomonas (16.2 %) were in the Bingöl, Fadlum, Tuzlagözü, and Kemah salterns, respectively (Figs.7, 8). Shannon–Wiener indices, rarefaction curves, and ACE and Chao1 richness estimators were used to characterize the diversity and species richness of the communities (Table 3). While the microbial communities of Fadlum and Tuzlagözü exhibited higher diversity (H′) and estimated richness (ACE, Chao1), Bingöl had the least diverse communities. Rarefaction analysis and the value of coverage (0.89) indicated that the sampling was not sufficient to predict the actual archaeal species richness of Fadlum, in which the most diverse archaeal community was detected (H′ = 4.83) (Table 3; Fig. 9).

Comparison of the four salterns according to cultivation and pyrosequencing results. Figures show shared genera between the four salterns (a), the genera that are unique to the salterns (b), and the other genera detected in some salterns (c)

Archaeal community compositions of crystallizers displayed as pie charts at genus-level (genera assigned at a 94.5 % sequence similarity cut-off value; genera representing less than 2 % of total archaeal communities are not shown)

Bacterial community compositions of the crystallizers are displayed as double pie charts. The inner circles indicates phyla, while the outer circles display genera assigned at a 94.5 % sequence similarity cut-off value (genera representing less than 2 % of total bacterial communities are not shown)

Rarefaction curves generated from pyrosequencing data for each sample (OTU definition based on 97 % sequence similarity)
Discussion
Tuzlagözü, Fadlum, Bingöl, and Kemah salterns could be classified as thalassohaline environments, and consist of ponds fed by hypersaline springs. Total salt concentration of the sampling ponds (20–27 %) was slightly lower than those previously studied, for instance, Maras (30–31 %), Sfax (20–36 %), and Santa Pola (22.4–37 %) (Antón et al. 2000; Maturrano et al. 2006; Boujelben et al. 2012). Total cell counts in the ponds varied between 3.8 × 106 and 2.0 × 107 cells mL−1. Similar results were reported before from other salterns and salt lakes using DAPI counts (Antón et al. 2000; Maturrano et al. 2006; Mutlu et al. 2008; Boujelben et al. 2012; Makhdoumi-Kakhki et al. 2012). The proportion of Archaea to Bacteria in the ponds was highly variable, but generally, the archaeal cells outnumbered the bacterial cells, as reported before (Maturrano et al. 2006; Mutlu et al. 2008; Boujelben et al. 2012; Makhdoumi-Kakhki et al. 2012).
A vast number of the archaeal OTUs (~96 %) detected in the pyrosequencing data showed high-sequence similarity with known archaeal genera and have been previously cultured or detected within clone libraries from solar salterns sand salt lakes (Burns et al. 2004; Walsh et al. 2005; Baati et al. 2008, 2010; Dillon et al. 2013). However, the number of novel archaeal genera (19) was notable, even though they comprised small fractions overall of the archaeal communities in our sampling sites. All archaeal sequences fell into the Class Halobacteria, and the phylotypes, assigned to a new family within the order Halobacteriales, constituted lower proportion of the overall archaeal sequences (1.2 %), and had similarity to the phylotypes obtained from the crystallizers in Australia (Oh et al. 2010). Analysis of the average community composition of the four solar salterns showed that the most highly represented genus was Haloquadratum which made up 23.6 % of the total archaeal pyrosequencing reads, followed by Halorubrum (19.6 %), Haloarcula (16.9 %), and Halonotius (16.6 %). OTUs affiliated with Haloquadratum, retrieved in this study, were similar to those obtained from clone libraries recovered from solar salterns in Tunisia, Mexico, and Spain (Baati et al. 2008; Dillon et al. 2013; Fernández et al. 2014). Halonotius-related pyrosequencing reads shared similarities with Halonotius pteroides isolated from Australian crystallizers and clones recovered from salt crystals processed from Mediterranean seawater (Burns et al. 2004; Baati et al. 2010). Strains belonging to genera Halorubrum and Haloarcula were isolated and related sequences detected by pyrosequencing indicating that it comprised a significant percentage of the archaeal portion of the communities. Another major group within the isolates was Halobacterium, for which associated OTUs constituted only a minor portion (0.6 %) of the overall pyrosequencing reads. When comparing our pyrosequencing data with those obtained from Santa Pola, in which microbiota inhabiting ponds of different salinities (13, 19, 37 %) were investigated, Haloquadratum was the most represented archaeal genus in the ponds of Bingöl (25 % salinity) and Santa Pola (37 % salinity). The abundance of OTUs related with Halorubrum and Haloquadratum were notable within the ponds of Tuzlagözü (20 % salinity) and Santa Pola (19 % salinity) both of which also contained Halomicrobium-, Natronomonas-, Halobacterium-, and Haloferax-related OTUs which constituted a lower proportion of the pyrosequencing reads (Ghai et al. 2011; Fernández et al. 2014).
Members of the Bacteroidetes phylum were found to be the major inhabitants of the crystallizers. Almost half of the bacterial OTUs were clustered within the Bacteroidetes (48.3 %) phylum in our study, and the sequences belonging to this phylum were similar to those previously retrieved from other hypersaline environments using clone libraries (Sørensen et al. 2005; Caton and Schneegurt 2012). The best known representative Salinibacter ruber, which is typically found in solar salterns and salt lakes, dominated Tuz Lake and the Santa Pola saltern (Spain) (Antón et al. 2002; Mutlu et al. 2008; Zhaxybayeva et al. 2013), was also isolated from the crystallization ponds of Bingöl and Tuzlagözü salterns. The most highly represented bacterial OTUs were also related to genera Salinibacter (23.2 %). OTUs belonging the phylum Bacteroidetes, accounting for ~10 % of the total, did not match with known genera. Some of the isolates clustered in phylum Bacteroidetes were obtained from the crystallization ponds and spring waters (excluding Kemah saltern), and 16S rRNA gene sequences of these isolates had <90 % sequence similarity to Aliifodinibius roseus and Aliifodinibius sediminis isolated from a salt mine in China (Wang et al. 2013). Some other isolates obtained in this study, within the Proteobacteria, could be also novel; and distantly related with genera Parvularcula and Marinobacter. Flavobacteriaceae-related sequences, assigned a new genus, comprised 3 % of the bacterial phylotypes, and related phylotypes obtained from the upper layer of the crust samples from a saltern in Eilat, Israel (Sørensen et al. 2005). The pyrosequencing reads related with Cyanobacteria were similar to those recovered from Guerrero Negro (Mexico) and Great Salt Plains (Harris et al. 2013; Caton and Schneegurt 2012). Almost all of the cyanobacterial sequences were within the order Chroococcales and showed similarity to sequences from the unicellular cyanobacterium Euhalothece, previously isolated from a hypersaline pool in Guerrero Negro and an evaporation pond in Eilat, Israel (Garcia-Pichel et al. 1998).
Kemah showed a very distinct microbial community profile among the four solar salterns investigated here, characterized by a dominance of bacterial cells, specifically members of the Proteobacteria that were revealed by FISH and pyrosequencing. Bacterial sequences recovered from Kemah were related to the genera Pseudomonas, Serratia, Stenotrophomonas, Sphingobium, Arthrobacter, and Acinetobacter (Fig. 8). Although some representatives of these genera have been previously recovered from saline environments (Jiang et al. 2007), they are not common inhabitants of solar salterns and are not typically considered to be halophilic. Salinibacter-related sequences constituted 6.4 % of the bacterial community in the crystallization pond of Kemah and no Haloquadratum-related sequences were detected. The possible cause of these differences in the microbial community composition of Kemah might be associated with anthropogenic impacts, but requires further investigation.
In this study, measures of alpha diversity varied between 2.63 and 4.83 (H′) for archaeal communities and 2.91 and 4.52 (H′) for bacterial communities. Calculation of these diversity indices was based on pyrosequencing data and yielded higher diversity values than those calculated using cloning data in previous studies (also based on a 3 % OTU distance level). For example, diversity indices based on clone libraries and DGGE bands in previous studies (Benlloch et al. 2002; Pašić et al. 2005; Baati et al. 2008; Caton et al. 2009; Oh et al. 2010; Boujelben et al. 2012) ranged between 1.64 and 2.10 (H′) for archaeal communities of crystallizers (34 % salinity) in Australia (Oh et al. 2010). Indices for archaeal communities from Secŏvlje solar salterns (Slovenia) were estimated to be 2.24 (H′) (Pašić et al. 2005). Alpha diversity indices for bacterial and archaeal communities in ponds of different salinities (18, 37, 38 % salinity) in the Baja solar saltern were 2.0, 1.1, and 2.5 (H′) and 0.6, 1.8, and 1.6 (H′) (respectively) (Dillon et al. 2013).
We were unable to detect members of the Nanohaloarchaea in our study and Narasingarao et al. (2012) has previously indicated that widely used (standard) 16S rRNA-targeted primer sets may not be suitable to reveal the presence of them. More recently, however, Gomariz et al. (2015) were able to obtain nanohaloarchaeal phylotypes using denaturing gradient gel electrophoresis of PCR-amplified fragments of 16S rRNA genes, indicating that PCR primer design can be optimized for this archaeal group.
In conclusion, the distribution of archaeal and bacterial groups in the four solar salterns were diverse, with the communities mainly composed of members of the Euryarchaeota, Bacteroidetes, Proteobacteria, and Cyanobacteria phyla. Representatives of the genera Haloquadratum and Salinibacter were the main inhabitants in the ponds suggesting that common inhabitants of the crystallizer ponds have been well described in previous studies. In addition, although the uncharacterized archaeal and bacterial taxa comprised relatively a small percentage of the overall population in our study, the number of novel genera detected displayed great diversity, suggesting that a more detailed examination needs to be performed to characterize them.
References
Aktimur HT, Tekirli ME, Yurdakul ME (1990) Geology of the Sivas-Erzincan Tertiary basin. Bull Miner Res Explor Inst Turkey 111:21–30
Amann RI (1995) In situ identification of micro-organisms by whole cell hybridization with rRNA-targeted nucleic acid probes. Molecular Microbial Ecology Manual. Kluwer academic Publishers, Dordrecht, pp 331–345
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925
Antón J, Llobet Brossa E, Rodríguez-Valera F, Amann R (1999) Fluorescence in situ hybridization analysis of the prokaryotic community inhabiting crystallizer ponds. Environ Microbiol 1:517–523
Antón J, Rosselló-Mora R, Rodríguez-Valera F, Amann R (2000) Extremely halophilic bacteria in crystallizer ponds from solar salterns. Appl Environ Microbiol 66:3052–3057
Antón J, Oren A, Benlloch S, Rodríguez-Valera F, Amann R, Rosselló-Mora R (2002) Salinibacter ruber gen. nov., sp. nov., a novel, extremely halophilic member of the Bacteria from saltern crystallizer ponds. Int J Syst Evol Microbiol 52:485–491
Baati H, Guermazi S, Amdouni R, Gharsallah N, Sghir A, Ammar E (2008) Prokaryotic diversity of a Tunisian multipond solar saltern. Extremophiles 12:505–518
Baati H, Guermazi S, Gharsallah N, Sghir A, Ammar E (2010) Microbial community of salt crystals processed from Mediterranean seawater based on 16S rRNA analysis. Can J Microbiol 56:44–51
Benlloch S, López-López A, Casamayor EO, Øvreås L, Goddard V, Daae FL, Smerdon G, Massana R, Joint I, Thingstad F, Pedrós-Alió C, Rodríguez Valera F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol 4:349–360
Boujelben I, Gomariz M, Martínez-García M, Santos F, Peña A, López C, Antón J, Maalej S (2012) Spatial and seasonal prokaryotic community dynamics in ponds of increasing salinity of Sfax solar saltern in Tunisia. Antonie Van Leeuwenhoek 101:845–857
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004) Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol 70:5258–5265
Casamayor EO, Massana R, Benlloch S, Øvreås L, Díez B, Goddard VJ, Gasol JM, Joint I, Rodríguez-Valera F, Pedrós Alió C (2002) Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4:338–348
Caton IR, Schneegurt MA (2012) Culture-independent analysis of the soil bacterial assemblage at the Great Salt Plains of Oklahoma. J Basic Microbiol 52:16–26
Caton TM, Caton IR, Witte LR, Schneegurt MA (2009) Archaeal diversity at the Great Salt Plains of Oklahoma described by cultivation and molecular analyses. Microb Ecol 58:519–528
Cifuentes A, Anton J, Benlloch S, Donnelly A, Herbert RA, Rodríguez-Valera F (2000) Prokaryotic diversity in Zostera noltii-colonized marine sediments. Appl Environ Microbiol 66:1715–1719
Çiner A, Koşun E (1996) Hafik Güneyindeki (Sivas Havzası) Oligo-Miyosen Yaşlı Çökellerin Stratigrafisi ve Sedimantolojisi. Turkish Assoc Pet Geol Bull 8:16–34
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM (2013) Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci 89:5685–5689
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469
Dillon J, Carlin M, Gutierrez A, Nguyen V, McLain N (2013) Patterns of microbial diversity along a salinity gradient in the Guerrero Negro solar saltern, Baja CA Sur, Mexico. Front Microbiol 4:399
Dyall-Smith M (2009) The halohandbook—protocols for haloarchaeal genetics, version 7.1. Martinsried, Germany, pp 12–15, 38–40. http://www.haloarchaea.com/resources/halohandbook/Halohandbook_2009_v7.1.pdf
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Fernández AB, León MJ, Vera B, Sánchez-Porro C, Ventosa A (2014) Metagenomic sequence of prokaryotic microbiota from an intermediate-salinity pond of a saltern in Isla Cristina, Spain. Genome Announc 2:e00045–e00114
Garcia-Pichel F, Nübel U, Muyzer G (1998) The phylogeny of unicellular, extremely halotolerant cyanobacteria. Arch Microbiol 169:469–482
Ghai R, Pašić L, Fernández AB, Martin-Cuadrado AB, Mizuno CM, McMahon KD, Papke RT, Stepanauskas R, Rodríguez-Brito B, Rohwer F, Sánchez-Porro C, Ventosa A, Rodríguez-Valera F (2011) New abundant microbial groups in aquatic hypersaline environments. Sci Rep 1:135
Gomariz M, Martínez-García M, Santos F, Rodriguez F, Capella-Gutiérrez S, Gabaldón T, Rosselló-Mora R, Meseguer I, Antón J (2015) From community approaches to single-cell genomics: the discovery of ubiquitous hyperhalophilic Bacteroidetes generalists. ISME J 9:16–31
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Harris JK, Caporaso JG, Walker JJ, Spear JR, Gold NJ, Robertson CE, Hugenholtz P, Goodrich J, McDonald D, Knights D, Marshall P, Tufo H, Knight R, Pace NR (2013) Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J 7:50–60
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319
Jiang H, Dong H, Yu B, Liu X, Li Y, Ji S, Zhang CL (2007) Microbial response to salinity change in Lake Chaka, a hypersaline lake on Tibetan plateau. Environ Microbiol 9:2603–2621
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S, Chun J (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62:716–721
Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR (1985) Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci 82:6955–6959
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Makhdoumi-Kakhki A, Amoozegar MA, Kazemi B, Pašić L, Ventosa A (2012) Prokaryotic diversity in Aran-Bidgol salt lake, the largest hypersaline playa in Iran. Microbes Environ 27:87–93
Maturrano L, Santos F, Rosselló-Mora R, Antón J (2006) Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol 72:3887–3895
Mutlu MB, Martínez-García M, Santos F, Peña A, Guven K, Antón J (2008) Prokaryotic diversity in Tuz Lake, a hypersaline environment in Inland Turkey. FEMS Microbiol Ecol 65:474–483
Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, Brocks JJ, Heidelberg KB, Banfield JF, Allen EE (2012) De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J 6:81–93
Nogales B, Moore ER, Abrham WR, Timmis KN (1999) Identification of the metabolically active members of a bacterial community in a polychlorinated biphenylpolluted moorland soil. Environ Microbiol 1:199–212
Oh D, Porter K, Russ B, Burns D, Dyall-Smith M (2010) Diversity of Haloquadratum and other haloarchaea in three, geographically distant, Australian saltern crystallizer ponds. Extremophiles 14:161–169
Oren A (2002) Halophilic microorganisms and their environments. Kluwer Academic Publishers, Dordrecht, p 393
Oren A (2008) Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst 4:13
Oren A, Rodrı́guez-Valera F (2001) The contribution of halophilic Bacteria to the red coloration of saltern crystallizer ponds. FEMS Microbiol Ecol 36:123–130
Pašić L, Bartual SG, Ulrih NP, Grabnar M, Velikonja BH (2005) Diversity of halophilic Archaea in the crystallizers of an Adriatic solar saltern. FEMS Microbiol Ecol 54:491–498
Sabet S, Diallo L, Hays L, Jung W, Dillon JG (2009) Characterization of halophiles isolated from solar salterns in Baja California, Mexico. Extremophiles 13:643–656
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH (1997) Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63:2884–2896
Sørensen KB, Canfield DE, Teske AP, Oren A (2005) Community composition of a hypersaline endoevaporitic microbial mat. Appl Environ Microbiol 71:7352–7365
Walsh DA, Papke RT, Doolittle WF (2005) Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol 7:1655–1666
Wang YX, Liu JH, Xiao W, Ma XL, Lai YH, Li ZY, Ji KY, Wen ML, Cui XL (2013) Aliifodinibius roseus gen. nov., sp. nov., and Aliifodinibius sediminis sp. nov., two moderately halophilic bacteria isolated from salt mine samples. Int J Syst Evol Microbiol 63:2907–2913
Zhaxybayeva O, Stepanauskas R, Mohan NR, Papke RT (2013) Cell sorting analysis of geographically separated hypersaline environments. Extremophiles 17:265–275
Acknowledgments
We would like to thank Prof. Dr. Mike Dyall-Smith for his helpful and critical comments on this work. The authors would like to thank Dr. Dionysios Antonopoulos from the Argonne National Laboratory (USA) for the English language review. This work was supported by the Anadolu University Research Foundation [Grant Number 1109F153].
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Oren.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Çınar, S., Mutlu, M.B. Comparative analysis of prokaryotic diversity in solar salterns in eastern Anatolia (Turkey). Extremophiles 20, 589–601 (2016). https://doi.org/10.1007/s00792-016-0845-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-016-0845-7