
Abstract
In this study, fluorescence in situ hybridization (FISH) and PCR-amplified fragments of the 16SrDNA gene were used to determine prokaryotes diversity in Urmia Salt Lake. Prokaryote cell population in Urmia lake range from 3.1 ± 0.3 × 106, 2 ± 0.2 × 108, 4 ± 0.3 × 108, and 1.8 ± 0.2 × 108 cells ml−1 for water, soil, sediment, and salt samples by DAPI (4́, 6-diamidino-2-phenylindole) direct count, respectively. The proportion of bacteria and archaea in the samples determinable by FISH ranged between 36.1 and 55% and 48.5 and 55.5%, respectively. According to the DGGE method, some bands were selected and separated from the gel, then amplified and sequenced. The results of sequences were related to two phyla Proteobacteria (16.6%) and Bacteroidetes (83.3%), which belonged to four genera Salinibacter, Mangroviflexus, Pseudomonas, and Cesiribacter, and the archaeal sequences were related to Euryarchaeota phyla and three genera Halonotius, Haloquadratum, and Halorubrum. According to our results, it seems that prokaryotic populations in this hypersaline environment are more diverse than expected, and bacteria are so abundant and diverse and form the metabolically active part of the microbial population inhabiting this extreme environment. Molecular dependent and independent approaches revealed a different aspect of this environment microbiota.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Extreme habitats as harsh conditions offer a unique chance to assess the types of microorganisms and further our understanding of growth parameters and/or requirements. In extreme habitats such as Hypersaline or saline lake, haloarchaea are predominated [1]. Previous researchers determined that domains archaea and bacteria are very important in hypersaline conditions [2]. The prokaryotes diversity hypersaline environments can be studied using genetic tools needless culture organisms [3]. The diversity of prokaryotes could not be investigated without the use of various methods to determine the characteristics of microorganisms. Microorganisms with different morphologies, physiologies, and phylogenetics were identified using various methods. The two techniques included are culture-dependent and culture-independent [4].
In modern environmental laboratories, molecular biology techniques replace traditional microbiological methods such as morphological characteristics, pathogenicity, nutritional requirements, or ecological niches. The disadvantage of these methods is that it is impossible to isolate pure cultures and cultivate some kinds of bacteria in artificial settings [5]. Recently, for investigation of microbial ecology, culture-independent molecular approaches by PCR-based methods are used [6]. However, the extraction of environmental DNA has dramatically improved without longer the limiting factor. In hypersaline environments, the discovery of organisms and unique genes is more interesting [3]. The method of molecular microbiology (FISH) has been applied for the evaluation of biodiversity. The techniques included especially staining and counting of prokaryotes using fluorescent-labeled oligonucleotide probes [7]. The halophilic/halotolerant microorganisms can be used in biotechnology; therefore, the researcher has preferred evaluation of biodiversity of hypersaline habitats [8,9,10,11,12]. The halophilic prokaryotes were applied in biotechnology and industry. Moreover, halophilic microorganisms can produce new biomaterials [13]. The microbial population of various hypersaline environments in Iran needs to be investigated. Urmia Lake is the 20th largest lake in the world located in northwest Iran. There are various published reports about Urmia Salt Lake [14,15,16,17]. This paper provided a valuable opportunity to advance the understanding of prokaryotic diversity in Urmia Salt Lake. However, the samples including soil, salt, and sediment were collected from the lake in Iran. The microbial communities were determined by FISH and DGGE of PCR-amplified fragments of 16S rRNA genes analysis.
Materials and Methods
Site Description
Lake Urmia (37° 32′ N, 045° 43′ E) is second lake in the earth and an endorheic salt lake in Iran and after Great Salt Lake in the western USA. The lake is located between the two provinces of East and West Azerbaijan and west of the southern portion of the Caspian Sea. The lake was the largest lake in the Middle East and the sixth biggest saline lake in the world, with 5,200 km2 surface area, 140 km long, 55 km wide, and 18 m deep. Depending on different conditions of space and time, the mean salinity is about 220 to 300 g/l (Fig. 1) [18].

Location of Urmia Salt Lake, in the northwest of Iran at Azerbaijan region. The arrow denotes the sampling sites (A, B, C, D, E, F, G, and H)
Sampling and Analysis
We collected 50 ml water and 50 g soil and salt from each site of Urmia Lake, stored at − 80 °C for molecular analysis, and transported to the laboratory within 24 h followed by pH and temperature measurements. The samples were stored at 4 °C for analysis. The pH and salinity of the samples were estimated using seven Multi-dual meter pH/conductivity (Mettler Toledo, Greifensee, Switzerland).
Fluorescence In situ Hybridization (FISH)
Microorganisms of samples (water, saline soil, sediment, and salt) were counted using DAPI (4′,6-diamidino-2-phenylindole) staining. The FISH technique was also examined using probes Arch915 [19] and EUB338 [20] for the archaea and the bacteria domains, respectively, in the samples.
Sample Preparation for In situ Hybridization
Water samples At first, a water sample was fixed by 4% final concentration of paraformaldehyde solution (54 ml of 37% (w/v) for 10 ml water), and then the sample was blend, and the suspension was kept at 4 °C for one night. The fixed samples were filtered by Millipore filter (0.22 μm) and then it was washed with distilled water (In this step, the filters can be kept for up to one month in a freezer at − 20 °C).
Saline soil and sediment The small parts of plants, rocks, roots, and insects were removed from samples (saline soil, sediment, and salt) with a sieve (2 mm mesh).
Then 4 g of each sample was added to 10 ml of paraformaldehyde solution (4% final concentration), mixed up thoroughly, and the suspension was stored at 4 °C for one night. The mixture was washed twice with 1X PBS, centrifuged at 10,000g for 5 min at 4 °C after each washing, and stored in PBS/ethanol (1:1) at − 20 °C for further processing.
Then, 4 ml PBS/ethanol was appended to 1 ml of the fixed sample and dispersed using ultrasound with an ultrasonic probe at minimum power (Sonopuls HD2200, Bandelin, Berlin, Germany) for 20 s utilizing one sonication pulse. The soil samples were also sonicated twice for 30 s by the same energy and pulsing rate as well as interval of 30 s. The sample was filtered on a Millipore filter (0.22 μm). In this stage, the filters can be stored for up to one month in a freezer at − 20 °C.
The filter was immersed in 100 µl of lysozyme (10 mg/g) and then incubated at 37 °C for 1 h and washed with distilled water. From this point on, the experiment was performed in darkness.
The filter was impregnated with 8 µl of the hybrid buffer and then 2 µl (5 ng/µl) of the probe was added to it and mixed carefully. It was stored in a moist Falcon at 48 °C for 2 h. Afterward, the filter was put into the washing buffer at 48 °C for 5 min. The process of washing was performed twice and then the filter was stained with DAPI. It was rinsed in water, then in 70% and 96% alcohol, air-dried, and fixed as a slide. The slides were kept in darkness at 4 °C until the desired time. However, the samples were evaluated using a fluorescence microscope (Olympus DX 59, Japan).
Extraction of Metagenomic DNA and Amplification of 16S rRNA Genes
Metagenomic DNA from the water sample was prepared as described previously [21, 22], and environmental DNA from salt, saline soil, and sediment were prepared using Jookar Kashi (2016) method [23].
DGGE was Carried Out for Amplifying 16S rRNA Genes
DGGE was investigated with the DCode System (Bio-Rad, Hercules, CA, USA), as Mutlu et al. (2008) reported previously [24]. The primers of DGGE analysis were the GC clamp 341F (5′-CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCCCTACGGGAGGCAGCAG) and 344F (5′-CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCC-ACGGGGCGCAGCAGGCGCGA) for bacteria and archaea, respectively, and 907R (5′-CCGTCAACCTTTRAGTTT-3′) used for both domains [25].
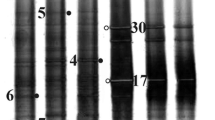
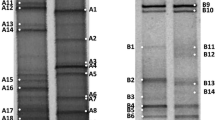
According to this method, the fragment of 16S rRNA genes of metagenomics was amplified with these primers. Then the PCR product was electrophoresed with bis-acrylamide gel with a linear gradient of denaturing agents and stained with ethidium bromide. Furthermore, the selected bands were separated, reamplified, and sequenced. The PCR procedure was 94 °C for 4 min, followed by 30 cycles of 94 °C for 50 s, 50 °C for 50 s, and 72 °C for 1 min with final 5-min extension at 72 °C. The products of PCR were sequenced using ANI 3730XL DNA sequencer at Macrogen (Seoul South Korea). The bands separated from DGGE were sequenced directly.
The NCBI GenBank database was designed to detect the reference 16SrDNA sequence using BLASTn and through the EzBioCloud Server (http://eztaxon-e.ezbiocloud.net) [26].
Phylogenetic analysis of strains and similar strains was performed by Clustal [27], Bioedit [28], and Mega 6 [29], and the phylogenetic tree was drawn for strains and similar strains using maximum likelihood and neighbor-joining methods [30].
Accession Numbers
The accession number was determined for the sequences in the GenBank sequence database (www.ncbi.nlm.nih.gov), and accession numbers were KJ634427-KJ634460.
Results and Discussion
Urmia Lake is the largest saline lake in the earth. After Dead Sea, this lake is a hypersaline lake with an active food. The previous results demonstrated that the lake is athalassohaline, and dominant ions were Cl− and Na+. Also, other ions included were Fe2+, K+, Mg2+, Ca2+, HCO3−, and Ca2+ [14, 31]. All the samples had a pH neutral, ranging from 7 to 7.5. The average temperature of the lake was from 20 to 25 °C, and the range of salinity of the samples indicated between 23 and 32% total salts. The total cell population was determined in the lake by DAPI. The total number of cells taken from water, soil, sediment, and salt samples were 3.1 ± 0.3 × 106, 2 ± 0.2 × 108, 4 ± 0.3 × 108, and 1.8 ± 0.2 × 108 cells ml−1, respectively. Previous research findings of Urmia Salt Lake reported live counts cells of water, soil, sediment, and salt, and (4.7 × 104–3.2 × 105), (5.6 × 105–6.5 × 106), (4.5 × 104–6.6 × 106), and (1.4 × 102–2.2 × 105) CFU ml−1 were obtained in culture media [32]. The population of bacteria and archaea in the lake determinable using the FISH test was between 36.1 and 55%, and 48.5 and 55.5%, respectively. The FISH technique demonstrated the estimated archaea of water, soil, sediment, and salt samples to be 51.61%, 48.5%, 45%, and 55.5, respectively. Moreover, the number of archaea in water, soil, sediment, and salt samples were 1.6 ± 0.2 × 106, 9.7 ± 0.2 × 107, 1.8 ± 0.4 × 108, and 1 ± 0.2 × 108, respectively. The bacterial cells detected in the water, soil, sediment, and salt samples were 45.16%, 55%, 52.5%, and 43.8%, respectively. Also, the results showed that bacterial cells in water, soil, sediment, and salt samples obtained were 1.4 ± 0.2 × 106, 1.1 ± 0.23 × 108, 2.1 ± 0.4 × 108, and 7.9 ± 0.3 × 107, respectively.
The FISH technique is applied widely and has many usages, but this test has limitations. Counting bacteria using the FISH test and general probes rarely yield results similar to using other technologies such as labeling nucleic acids. The possibility of the identity of the target cell is in the range of 0 to 100% with this test. Total cell counts in the lake determined are in the range of 108 cells ml−1, more or less similar to that determined from other hypersaline communities [33, 34], but others reported the microbial cell densities of 106 cells ml−1 [35, 36].
The molecular methods including culture-dependent and culture-independent approaches were used for analyzing bacterial community. The moderately and extreme halophilic prokaryotes were identified by molecular and microbiological methods in a wide range of these saline habitats. The different types of organisms and their relative abundance in the community were identified as biodiversity. The diversity of environments was studied by various molecular methods and DNA extracted from samples. Moreover, unculturable bacteria were determined via molecular methods in the laboratory. PCR-DGGE technique provided information about numerical changes in the numerically dominant bacterial populations. The FISH method could be used to investigate the taxon composition of prokaryote communities by phylogenetic probes [37]. The previous research showed that just 1% of all prokaryotes could be determined and isolated on culture media in the laboratory [4].
The DGGE fingerprints of the sampled were analyzed, and 33 different bands (18 bacteria and 15 archaea) were determined. The phylogenetic analysis indicated that bacteria belonged to two phyla (6%) (Table 1). The phylogenetic tree for archaea and bacteria was prepared by the maximum likelihood method, and the strains on the tree are identified in Fig. 2a and b.

a Archaeal phylogenetic tree of a partial 16SrDNA gene sequence derived from the DGGE bands. The tree has been rooted in Methanocalculus chunghsingensis K1F9705bT (AF347025). b The phylogenetic tree of bacterial isolates was derived from the DGGE bands. The sequence Rubritalea tangerina YM27-005T (AB297806.1) was applied as an outgroup
The DNA environment was isolated from the samples and applied for the amplification fragments of 16SrDNA and a fragment of the bop gene by PCR, cloning, and sequencing. The clone libraries of archaeal library related to five genera, including Halonotius, Halolamina, Haloquadratum, Halomicroarcula, and Halorhabdus [15]. The results determined the bacteria belonged to four phyla (Bacteroidetes, Cyanobacteria, Actinobacteria, and Firmicutes) and six genera, including Acaryochloris, Adhaeribacter, Brachybacterium, Gloeocapsopsis, Cesiribacter, BacillusBacillus. The two genera of archaea (Halonotius, Haloquadratum) and one genus of bacteria (Cesiribacter) were common to both studies. However, the clone libraries of bop gene identified four genera Halorubrum, Natrialba, Haloquadratum, and Natrinema. The results of bop phylogeny were related to the 16SrDNA phylogeny [15].
In another research in 2014, water, soil, and salt samples were collected from the east of the lake and bacterial isolated via the culture-dependent method. These isolated strains belonged to Bacillus, Thalassobacillus, Planococcus, Virgibacillus, Ornithinibacillus, Halomonas, Pseudomonas, Providencia, Salicola, Psychrobacter, Terribacillus, Staphylococcus, Oceanobacillus, and Planomicrobium. The genera Pseudomonas and Bacillus were common to our results [14].
Moreover, in another study, the biodiversity of the lake was determined using conventional culture-dependent methods. The samples (water, soil, sediment, and salt) were obtained from east and western sites in Urmia Salt Lake. The samples were cultured on MGM, MH, and SWN medium by direct plating, dilution plating, and long incubation period. Two hundred and twenty-eight microorganisms were obtained. Of these, 36 isolates were applied for sequencing and phylogenetic analyses. The findings of their report determined 36 strains which represented 8 species, belonging to 3 genera of archaea (Halorubrum, Haloarcula, Haloterrigena). As a total, bacterial isolates are related to Salicola and Pseudomonas. All strains demonstrated 96.5 to 100% similarity in 16SrDNA sequencing. Of these, five strains showed less than 98.7% sequence similarity to the closest known strains and were representatives as new taxa of Urmia Lake [32].
However, there was a low overlap between the results obtained in this research and other findings of the lake because the low number of strains were sequenced by the DGGE technique. Therefore, we can increase the number of samples for obtaining acceptable results. A comparison of our results and other culture-dependent and culture-independent studies suggests that culture-independent approaches, such as oligonucleotide microarrays and sequencing 16SrDNA genes from DGGE and clone libraries also, and culture-dependent methods are necessary for identifying of diversity.
Based on the results of this study, we suggested that other media can be used to cultivate halophilic and halotolerant bacteria from hypersaline ecosystems. Also, sequences from the culture-dependent approach of other study and molecular approaches used in the present study are different. It could be concluded that the different methods applied to investigate the microbial diversity of one ecosystem may result in various outcomes. For the study of microbial ecology, it is necessary to obtain DNA from the samples. The microbial communities can be determined by this method. The extraction of DNA from soils and sediments is difficult. Many methods of DNA extraction were applied for molecular analysis [23].
Our results determined that Urmia Lake is a critical extreme habitat for further study using combined various culture methods including culture media type and growth conditions and other molecular techniques. We collected saline soil, and sediment samples contain high clay and organic matter. The clay and organic matter adsorbed DNA. Furthermore, the extraction of DNA from microorganisms was not very successful. However, molecular methods exhibited microorganisms that are commonly determined in extreme environments; a better comparison between culture-dependent and culture-independent approaches would require a multitude of isolates grown under variable conditions. There are many hypersaline ecosystems in Iran, only a few of which have been studied for their microbial community composition.
The results of DGGE in Aran-Bidgol Salt Lake showed that archaeal sequences belonged to the genera Halorubrum, Haloquadratum, Halonotius, and Haladaptatus, and bacterial sequences related to Halorhodospira. The result determined overlaps between two salinity sites and Halorubrum, Haloquadratum, and Halonotius were common, but there is no overlap between the results of the bacterial sequence [38].
Didari et al. reported culturable diversity of halophilic and halotolerant bacteria in Aran-Bidgol Lake. Their results showed that the isolates belonged to genera Bacillus, Halomonas, Oceanobacillus, Salinicoccus, Thalassobacillus, Ornithinibacillus, Halobacillus, Salicola, Virgibacillus, Aerococcus, Arthrobacter, Idiomarina, Parageobacillus, Staphylococcus, Acinetobacter, Aneurinibacillus, Brevibacillus, Brevundimonas, Chromohalobacter, Gracillibacillus, Jeotgalicoccus, Kocuria, Marinilacibacillus, Marinobacter, Microbacterium, Panenibacillus, Paracoccus, Piscibacillus, Pseudomonas, and Sediminibacillus. According to their results, most strains were not identified in our study [39].
In a similar research, phylogenetic analysis of Bohai Bay Solar Saltworks, China, was studied using the DGGE methods. They found that bacteria belonged to γ-proteobacteria (34%), Firmicutes (14%), and Bacteroidetes (9%) [40].
In another study by Ahmad et al., archaeal diversity in salt pan sediment from Mumbai, India, was reported using 16SrDNA-dependent molecular phylogeny. They found that archaea belonged to Crenarchaeota and Euryarcheaota [41].
Shuaibing et al. studied the diversity of archaea in Ebinur Lake wetland, China, by 16SrDNA cloning library. They showed that archaea belonged to Euryarchaeota, Thaumarchaeota, and Crenarcheota. They reported that soil moisture, electrical conductivity, soil organic matter content, and pH affect archaea diversity [42].
The diversity of archaea and bacteria was evaluated using the cultivation method in Sambhar Salt Lake, India. The results showed that predominant groups of bacteria and archaea were Alkalibacillus and Natronococcus [43].
Next-generation sequencing is one of the methods that can be used to analyze microbial diversity in extreme habitats. The halophilic microbial diversity of the Karak salt mine, Pakistan, was studied using 16SrDNA Illumina amplicon sequencing. The pH and salinity of the Karak salt mine were 7.14 and 32%, respectively. The results showed that the prokaryotic community belonged to bacteria (60%) and archaea (22%) with the phyla members Euryarchaeota, Bacteroidetes, Proteobacteria, Firmicutes, and Acetothermia [44].
Derui et al. have assessed bacterial diversity in Qaidam Basin, China. The 16SrDNA gene Illumina sequencing analyses determined that the dominant phyla were Proteobacteria, Firmicutes, Bacteroidetes, and Deinococcus-Thermus [45].
Shaoxing et al. studied prokaryotic diversity in salt lakes, solar salterns, and salt mines. They used clone library and Illumina Miseq sequencing. Moreover, high-throughput sequencing results determined that proteobacteria, Firmicutes, and Bacteroidetes were the phyla dominant within hypersaline habitats [46]
The microbial diversity of Chagan Lake, China, was studied using 16SrDNA-targeted metagenomics analysis with Illumina HiSeq 2500. Based on results, 48 bacterial phyla were found, and the dominant phyla were Proteobacterium, Actinomycetes, and Bacteroides [47].
Different methods can be used to study biodiversity, which has its advantages and disadvantages. Simultaneous use of several methods can provide complete and better results. The cultured-dependent approach can estimate 1% of microbial diversity. Moreover, cultured-independent 16SrDNA gene sequencing increases our knowledge of biodiversity. DGGE is a valuable method to evaluate microbial consortium [48]. This method prepares valuable information, including assessing microbial communities, analyzing population changes, sequence heterogeneities, and taxonomic database [49].
The results of DGGE were sequenced using Sanger sequencing with no/minimal mistakes and identified the dominant populations. However, there are limitations to DGGE as the only method of identifying diversity. The disadvantage of DGGE is that multiple copies determine one species. Moreover, Sanger sequencing is expensive and cannot identify rare microorganisms. In other methods such as next-generation sequencing, thousands to millions of sequences are identified in one test, which costs less than the Sanger method. The NGS does not require the preparation of a clone library and electrophoresis. Further, preparation of NGS libraries is accomplished in a cell-free system; the sequence output is directly identified. Moreover, it prepares shorter readings with a higher error rate than the Sanger sequence. Ion Torrent method as NGS in comparison to DGGE identified short read length (100–200 bp). The phylogenetic identification of shorter sequence size (100 bp) is not successful and may not be sufficient for taxonomic identification [50].
Finally, climate change for biodiversity and ecosystems is a serious threat. Microbial species and other organisms in a habitat are affected by climate change. Climate change and human activities have largely affected Urmia Lake. In the last two decades, an increase of the dryness and the salinity and a decrease of the surface area of the lake affected biodiversity [51].
Conclusion
In this study, the samples of water and soil were collected from Urmia Salt Lake. The prokaryotes’ diversity was determined using the FISH technique and the DGGE method. The results showed that the two methods possess a functional potential for distinguishing prokaryotes diversity. These techniques are simple, fast, and generally utilized as a suitable tool for the first step of prokaryotes’ diversity. Our findings found that Urmia Lake is an extreme ecosystem that should combine various culture-dependent and culture-independent methods for further study. Finally, this research fills the gaps in our understanding of prokaryotic diversity in Urmia Salt Lake. Moreover, there is a significant difference in microbial diversity in different places of Urmia Lake. Therefore, it is necessary to conduct further research with other methods. Future research using next-generation sequencing methods will expand our knowledge of Urmia Lake as a saline environment.
Change history
21 July 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00284-021-02614-6
References
Boutaiba S, Hacene H, Bidle KA, Maupin-Furlow JA (2011) Microbial diversity of the hypersaline Sidi Ameur and Himalatt Salt Lakes of the Algerian Sahara. J Arid Environ 75(10):909–916
Hedi A, Essghaier B, Cayol JL, Fardeau ML, N, (2014) Prokaryotic biodiversity of halophilic microorganisms isolated from Sehline Sebkha Salt Lake (Tunisia). Afr J Microbiol Res 8(4):355–367
Oren A, Baxter BK, Weimer BC (2009) Microbial communities in Salt Lakes: Phylogenetic diversity, metabolic diversity, and in situ activities. Nat Resour Environ Issues 15(51):1–8
Ghiasian M, Akhavan Sepahy A, Amoozegar MA, Saadatmand S, Shavandi M (2017) Bacterial diversity determination using culture-dependent and culture-independent methods. Global J Environ Sci Manage 3(2):153–164
Karło A, Ziembińska A (2013) Modern techniques used for biodiversity analysis in bacterial environmental communities. CHEMIK 67(11):1105–1114
Shipeng Lu, Minjeong P, Hyeon-Su R, Dae Sung L, Woojun P, Jeon CO (2006) Analysis of microbial communities using culture-dependent and culture-independent approaches in an anaerobic/aerobic SBR reactor. J Microbiol 44(2):155–161
Eickhorst T, Tippkotter R (2008) Improved detection of soil microorganisms using fluorescence in situ hybridization (FISH) and catalyzed reporter deposition (CARD-FISH). Soil Biol Biochem 40:1883–1891
Dastgheib MM, Amoozegar MA, Khajeh K, Shavandi M, Ventosa A (2012) Biodegradation of polycyclic aromatic hydrocarbons by a halophilic microbial consortium. Appl Microbiol Biotechnol 95:789–798
Delgado-García M, Valdivia-Urdiales B, Aguilar-González CN, Contreras-Esquivel JC, Rodríguez-Herrera R (2012) Halophilic hydrolases as a new tool for the biotechnological industries. J Sci Food Agric 92(13):2575–2580
Llamas I, Amjres H, Mata JA, Quesada E, Béjar V (2012) The potential biotechnological applications of the exopolysaccharide produced by the halophilic bacterium Halomonas almeriensis. Molecules 17:7103–7120
Rohban R, Amoozegar MA, Ventosa A (2009) Screening and isolation of halophilic bacteria producing extracellular hydrolyses from Howz Soltan Lake. Iran J Ind Microbiol Biotechnol 36(3):333–340
Tang J, Ai-Ping SP, Bromfield JZ, Shuang-Cheng L, Shi-Quan W, Qi-Ming D, Ping L (2011) 16S rRNA gene sequence analysis of halophilic and halotolerant bacteria isolated from a hypersaline pond in Sichuan, China. Ann Microbiol 61:375–381
Safarpour A, Amoozegar MA, Ventosa, A (2018) Hypersaline environments of Iran: prokaryotic biodiversity and their potentials in microbial biotechnology. Extremophiles in Eurasian Ecosystems. pp 265–298
Jookar Kashi F, Owlia P, Amoozegar MA, Yakhchali B, Kazemi B (2014) Diversity of cultivable microorganisms in the eastern part of Urmia Salt Lake. Iran J Microbiol Biotech Food Sci 4(1):36–43
Jookar Kashi F, Owlia P, Amoozegar MA (2018) Evaluation of prokaryotic diversity in hypersaline environment by culture-independent method. Modares J Biotech 9(1):137–144
Zununi V, Forouhandeh S, Hassanzadeh H, Peter Klenk S, Hejazi H, Hejazi MA (2011) Isolation and characterization of halophilic bacteria from urmia lake in Iran. Microbiology 80(6):834–841
Mehrshad M, Amoozegar MA, Yakhchali B, Shahzade Fazeli A (2012) Biodiversity of moderately halophilic and halotolerant bacteria in the western coastal line of Urmia lake. Biol J Microorg 2:49–70
Farzin S, Ifaei P, Farzin N, Hassanzadeh Y, Aalami MT (2012) An investigation on changes and prediction of Urmia Lake water surface evaporation by chaos theory. Int J Environ Res 6(3):815–824
Stahl DA, Amann R (1991) Development and application of nucleic acid probes in bacterial systematics. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 205–248
Amann R, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925
Benlloch S, Acinas SG, Martínez-Mucia AJ, RodríguezValera F (1996) Description of prokaryotic biodiversity along the salinity gradient of a multipond solar saltern by direct PCR amplification of 16S rDNA. Hydrobiologia 329:19–31
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004) Combined use of cultivation-dependent and cultivation independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol 70:5258–5265
Jookar Kashi F (2016) An improved procedure of the metagenomic DNA extraction from saline soil, sediment and salt. Int Lett Nat Sci 60:38–45
Mutlu MB, Martínez-García M, Santos F, Peña A, Guven K, Antón J (2008) Prokaryotic diversity in Tuz Lake, a hypersaline environment in Inland Turkey. FEMS Microbiol Ecol 65:474–483
Muyzer G, De Waal EC, Uitterrlinden AG (1993) Profiling in complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Hall TA (1999) BioEdit: A user-friendly biological sequence alignment editor andanalysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:958
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6 (2013) Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–27299.
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39(4):783–791
Grant WD (2004) Life at low water activity. Phil Trans R Soc Lond 359:1249–1267
Jookar Kashi F, Owlia P, Amoozegar MA, Yakhchali B (2014) Culturable prokaryotic diversity of Urmia Salt Lake. Mod Genet 9(38):313–328
Guixa-Boixareu N, Calderón-Paz J, Heldal M, Bratbak G, Pedrós-Alió C (1996) Viral lysis and bacterivory as prokaryotic loss factors along a salinity gradient. Aquat Microbial Ecol 11:215–227
Ochsenreiter T, Pfeifer F, Schleper C (2002) Diversity of Archaea in hypersaline environments characterized by molecular-phylogenetic and cultivation studies. Extremophiles 6:267–274
Antón J, Llobet-Brossa E, Rodriguez-Valera F, Amann R (1999) Fluorescence in situ hybridization analysis of the prokaryotic community inhabiting crystallizer ponds. Environ. Microbiol 5:1517–523
Maturrano L, Santos F, Rossello-Mora R, Anton J (2006) Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol 72:3887–3895
Torsvik V, Lise Daae F, Sandaa RA, Øvreas L (1998) Novel techniques for analysing microbial diversity in natural and perturbed environments. J Biotechnol 664:53–62
Makhdoumi-Kakhki A, Amoozegar MA, Kazemi B, Pašic L, Ventosa A (2012) Prokaryotic diversity in Aran-Bidgol Salt Lake, the largest hypersaline playa in Iran. Microbes Environ 27(1):87–93
Didari M, Bagheri M, Amoozegar MA, Bouzari S, Babavalian H, Tebyanian H, Hassanshahian M, Ventosa A (2020) Diversity of halophilic and halotolerant bacteria in the largest seasonal hypersaline lake (Aran-Bidgol-Iran). J Environ Health Sci Eng 87:1–11
Jiaojiao Z, Guannan M, Yuangao D, Jinggang D, Van Gilbert S, Sui L (2016) Bacterial diversity in Bohai Bay Solar Saltworks. China Curr Microbiol 72:55–63
Nasier A, Sarika S, Farrah GK, Rajinder K, Sarojini J, Malik ZA, Ghulam NQ (2008) Phylogenetic analyses of archaeal ribosomal DNA sequences from Salt Pan Sediment of Mumbai. India Curr Microbiol 57:145–152
Shuaibing H, Jun T, Wenge H, Chao M (2019) Diversity of archaea and its correlation with environmental factors in the Ebinur Lake Wetland. Curr Microbiol 76:1417–1424
Swapnil K, Neelima D, Yogesh S, Avinash S (2020) Cultivation of diverse microorganisms from hypersaline lake and impact of delay in sample processing on cell viability. Curr Microbiol 77:716–721
Cycil LM, DasSarma S, Pecher W, McDonald R, AbdulSalam M, Hasan F (2020) Metagenomic insights into the diversity of halophilic microorganisms indigenous to the Karak Salt Mine. Pakistan Front Microbiol 11:1567
Derui Z, Rui H, Qifu L, Xiang G, Jiangwa X, Guoping S, Yongzhen L, Rong W (2020) An evaluation of the core bacterial communities associated with hypersaline environments in the Qaidam Basin. China Arch Microbiol 202(8):2093–2103
Shaoxing C, Yao X, Libby H (2020) Geographical isolation, buried depth, and physicochemical traits drive the variation of species diversity and prokaryotic community in three typical hypersaline environments. Microorganisms 8(120):1–14
Zhang L, CAI Y, Jiang M, Dai J, Guo X, LI W, LI Y. (2020) The levels of microbial diversity in different water layers of saline Chagan Lake. China J Oceanol Limnol 38:395–407
De Mandal S, Panda AK, Bisht SS, Kumar NS (2015) Microbial ecology in the era of next generation sequencing. Next Generat Seq Appl 21:1–6
Raquel AC, Victor HS, Gretty KV (2018) Microbial diversity assessment by PCR-DGGE analysis in national sanctuary of ampay in Perú. Adv Appl Microbiol 11(3):1–6
Samarajeewa AD, Hammad A, Masson L, Khan IUH, Scroggins R, Beaudette LA (2015) Comparative assessment of next-generation sequencing, denaturing gradient gel electrophoresis, clonal restriction fragment length polymorphism and cloning-sequencing as methods for characterizing commercial microbial consortia. J Microbiol Methods 108:103–111
Matthias S, Robert G, Sebastian T (2020) Environmental degradation at Lake Urmia (Iran): exploring the causes and their impacts on rural livelihoods. GeoJournal 8:1–15
Acknowledgements
The authors are grateful to Shahed University for supporting this work.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jookar Kashi, F., Owlia, P., Amoozegar, M.A. et al. Halophilic Prokaryotes in Urmia Salt Lake, a Hypersaline Environment in Iran. Curr Microbiol 78, 3230–3238 (2021). https://doi.org/10.1007/s00284-021-02583-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-021-02583-w