
Abstract
Wide-ranging changes to the coastline of East Asia during the glacial periods of the Quaternary might have significantly influenced the genetic structure of coastal plants. To verify its historical migration and potential breaks to gene flow, we investigated the genetic structure of the vulnerable halophyte Suaeda malacosperma, whose distribution is restricted to brackish tidal areas of Korea and Japan. Two chloroplast DNA (cpDNA) regions (rpl32-trnL, trnH-psbA) were used for five individuals each from 11 populations. SNP data obtained via multiplexed ISSR genotyping by sequencing (MIG-Seq) were used for 181 individuals from those populations. Ecological niche modeling (ENM) was combined with genetic analyses to compare current and past distributions. The cpDNA sequences and MIG-Seq data revealed largely congruent results indicating that S. malacosperma consists of three genetic clusters: western coast of Korea, southern coast of Korea, and Japan. The patterns produced through structure analysis and cpDNA haplotypes showed that the gene flow occurred at or after the last glaciation from Japan to the southern coast of Korea via the Korea/Tsushima Strait land bridge. Despite the coastal habitat of this species, ocean currents had less influence. Integrating genetic data with past distribution models by ENM provided insight into potential recolonization routes from refugia in each region. Those data suggested that, instead of contemporary gene flow, a historical range shift due to climate change affected the population structure. These results support the need for a conservation strategy and advance our understanding about the historical range dynamics of coastal plants in temperate East Asia.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coastal regions play roles in biodiversity and ecology (Costanza et al. 1997; UNEP 2006), because they are very productive areas and provide habitats for a wide array of organisms. The distribution of those organisms can be influenced by both natural and artificial factors. The biodiversity and ecosystems of coastal areas have suffered not only from the effects of shifting habitats, invasive species, and pollution (UNEP 2006), but also from repeated glacial periods during the Pleistocene by changes in climate and sea level (Clark et al. 2009). Among those factors, fluctuations of the sea level have had a critical impact on genetic diversity and the historical distribution of organisms according to regression and transgression of the oceans (Kadereit et al. 2005; Weising and Freitag 2007; Han et al. 2015).
In inland East Asia, climate change during glacial periods altered plant populations, as elucidated by putative refugia and corridors for migration (reviewed by Qiu et al. 2011). Land bridges that formed in response to climate change played an important role as corridors that shaped the contemporary genetic structure of those populations (Lee et al. 2013, 2016; Jin et al. 2016). Unlike inland plants, coastal plants are affected by both glaciation related changes and the dispersal mode through the ocean. That mode theoretically affects the genetic patterns of coastal plants across long distances and is considered an important factor when explaining their genetic diversity (Kadereit et al. 2005; Weising and Freitag 2007; Westberg and Kadereit 2009; Han et al. 2015). Although the migration process and historical refugia of inland plants in East Asia have been widely studied (Qiu et al. 2011) and despite their interesting biological implications, only few studies have focused on the phylogeography of coastal plants of that region (Han et al. 2015).
Suaeda malacosperma Hara is an annual halophyte that grows along the coastlines of Korea and Japan. The chloroplast genome of S. malacosperma was first reported in the genus Suaeda Scop. (Park et al. 2018). These plants were first recorded in and described from Japan (Hara 1942) and more recently discovered in salt-marshes of Korea (Shim et al. 2001). In contrast to other plants of coastal salt-marshes, as, e.g., S. maritima subsp. asiatica Hara and S. japonica Makino, it occurs only locally on brackish sites, preferably in estuaries. This species is included into the Red List (VU) in Japan (https://ikilog.biodic.go.jp/Rdb/booklist) and as “not evaluated” (NE) in Korea (National Institute of Biological Resources 2012). Several local extinctions have been reported in Japan (Nakanishi 2001). Because of its distribution to coastal areas, the species presents advantages for phylogeographic studies that examine the direct influence of changes in coastlines and the formation of land bridges during glacial periods. For example, after the Last Glacial Maximum (LGM: ca 19,000 years before present), wide continental shelves of the Yellow Sea and the East China Sea (ECS), as well as the Korea/Tsushima Strait land bridge, were submerged, and afterward the coastlines gradually moved inland over very long distances (Kimura 1996; Yoo et al. 2016).
Although their biological and ecological significance has been recognized since the 1950s (Costanza et al. 1997), large coastal regions and estuaries in Korea and Japan are just now transformed by reclamation efforts to increase arable lands (Suzuki 2003; Murray et al. 2014; Choi 2014) with inherent strong effects on the diversity of autochthonous organisms (Ryu et al. 1997; Sato 2006). These activities have increased the need to conserve S. malacosperma.
This study aims to (1) elucidate the genetic structure and potential geographic barriers of S. malacosperma over its complete range of distribution, (2) detect recolonization routes that resulted from a change in the coastline after the LGM, and (3) obtain genetic information that helps explain historic geographical processes and contemporary gene flow and can be used in establishing a conservation strategy for these coastal populations. In order to infer historical distribution change after the LGM, we applied the ecological niche modeling (ENM) that is utilized in phylogeographic studies to investigate climate change-associated correlations between distribution shifts and genetic structure (Carstens and Richards 2007; Qi et al. 2012; García-Fernández et al. 2017). Because the deposition of fossils or pollen grains is difficult in such coastal environments, a technique such as the ENM is the only way to model the historical distribution of a species. The genetic structure of S. malacosperma was analyzed using the multiplexed inter-simple sequence repeat genotyping by sequencing (MIG-Seq) (Suyama and Matsuki 2015) and chloroplast DNA (cpDNA) sequences. The MIG-Seq is a useful technique for analyzing the genetic structure by producing reduced representatives from an entire genome; it has recently been used in studies of population genetics and genetic ecology (Takahashi et al. 2016; Tamaki et al. 2017; Yoichi et al. 2018). Characteristics of cpDNA, such as maternal inheritance, non-recombination, and a substitution rate lower than that of nuclear DNA, could complement MIG-Seq analysis. In East Asia, phylogeographic studies of coastal plants are poorly performed compared to European coastal plants (Arafeh and Kadereit 2006; Kadereit et al. 2005; Jakob et al. 2007; Kadereit and Westberg 2007; Escudero et al. 2010; Prinz et al. 2013; García-Fernández et al. 2017). Data generated from our study using ENM, MIG-Seq and cpDNA provide a phylogeographical insight to coastal plants in East Asia.
Materials and methods
Sampling and DNA extraction
Leaves were collected from 181 individual plants belonging to 11 populations of S. malacosperma growing along the coastal regions of Korea and Japan (Table 1). The sampling covered almost the entire distribution area of the species. Each sample was dried in silica gel and stored at room temperature before genomic DNA was extracted using MG™ Plant SV (MGmed, Seoul, Korea).
Chloroplast DNA sequencing
PCR amplifications were performed for the two non-coding regions of cpDNA: rpl32-trnL (Shaw et al. 2007) and trnH-psbA (Sang et al. 1997). These regions were selected after preliminary screening of seven non-coding regions (trnL-trnF, trnH-psbA, psbB-psbH, atpB-rbcL, rbcL-atpB, rpl32-trnL, and rpl16 intron). Because of the expected low genetic diversity of each population in the process of development of microsatellite marker for S. malacosperma, we used 55 samples (five per population) of the 181 samples collected. We conducted PCR with a GeneAmp® PCR System 2720 Thermal Cycler (Applied Biosystems, Foster City, CA, USA). Each reaction mixture contained MG 2X Taq MasterMix without dye (Mgmed), ca. 10 ng of DNA, and 20 nM of primers in a total volume of 20 µL. Conditions included an initial denaturation at 95 °C for 5 min, followed by 30 amplification cycles comprising 95 °C for 1 min, 52 °C (55 °C for nrITS) for 1 min, and 72 °C for 1 min, with a final extension at 72 °C for 7 min. After the PCR products were visualized on 2% agarose gels, they were treated with a MG PCR Purification kit (MGmed), and sequenced with the ABI 3100 Genetic Analyzer, using the ABI BigDye™ Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). The sequences identified in S. malacosperma were deposited in the GenBank database (MH845066–MH845175).
SNP analyses by MIG-Seq
We used MIG-Seq to conduct genome-wide SNP analysis, constructing a MIG-Seq library of 181 samples according to a protocol slightly modified from that of Suyama and Matsuki (2015). Briefly, the annealing temperature for the first PCR was changed to 38 °C to obtain more amplicons, while the second PCR involved indexed forward and reverse primers. After this two-step process, the amplified PCR products were purified and quantified for sequencing on an Illumina MiSeq platform (Illumina, San Diego, CA, USA), using a MiSeq Reagent kit v3 (150 cycles; Illumina).
After obtaining the NGS data, we removed the adapter and anchor in the sequenced reads with a FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Low-quality and overlapping reads were removed with the FASTX Toolkit and TagDust (Lassmann et al. 2009). We then analyzed the filtered reads with Stacks v1.48 (Catchen et al. 2013). Each stack was piled by the ustacks program, setting the minimum depth coverage at m = 10 and the maximum distance at M = 1. Allowing for four mismatches, the cstacks program produced the loci catalogue of stacks while the sstacks program was used to match the catalogue with each sample. To minimize the risk of missing data and avoid having private loci in a population, we set the minimum percentage of a locus shared within a population at 75% (r = 0.75) and the minimum number of populations where a locus had to be present at p = 2. The data files for structure and the fasta format were exported with the population program, and the structure format was applied to only the first SNP per locus (–wrire_single_snp option) to avoid any linkages between SNPs.
Analysis of population genetics
We used the population program in stacks to estimate genetic diversity (percentage of polymorphic loci), expected heterozygosity (HE), observed heterozygosity (HO), nucleotide diversity (π), and Wright’s F-statistics. To analyze the genetic structure, we applied the Bayesian model-based clustering method (STRUCTURE 2.0; Pritchard et al. 2000), conducting 20 runs for each value of K from 1 to 12. Each run employed the parameter set which was configured to 200,000 iterations (burn-in), following 300,000 Markov chain Monte Carlo repetitions and admixture model with default setting. Data set and parameter set were deposited at Figshare (https://doi.org/10.6084/m9.figshare.7387046). The optimal number of clusters (K) was estimated by using the STRUCTURE HARVESTER (Earl and von Holdt 2012), according to the method of Evanno et al. (2005). We used the Clustering Markov Packager Across K (CLUMPAK) online program (Kopelman et al. 2015) to summarize the results from each run and to generate representative pie charts associated with each K value.
Ecological niche modeling
We modeled the potential distribution of S. malacosperma historically and currently, using Maxent 3.4.1 (Merow et al. 2013). Occurrence data for this species included sample localities from our study as well as data published earlier (Nakanishi 2001; Shim et al. 2009; Online Resource 1). We downloaded altitude and current/past climate data according to our 19 bioclimatic variables (Online Resource 2) from WorldClim Version 1.4 (http://www.worldclim.org/) (Hijmans et al. 2005). To reconstruct the previous potential distributions, we utilized three past climate models for LGM: the Community Climate System Model (CCSM4; Gent et al. 2011), Earth System Model based on the Model for Interdisciplinary Research On Climate (MIROC-ESM; Watanabe et al. 2011), and the Max Planck Institute for Meteorology Earth System model (MPI-ESM-P). Whereas the past climate data for the LGM had a 2.5-m arc resolution, the others were 30-s arc resolutions. To adapt the altitude of the LGM, we obtained the ETOPO1 Global Relief Model (https://doi.org/10.7289/V5C8276M). Because all data for each ENM required the same resolution, we re-sampled the data to make its resolution the same as that of the LGM climate and modified it to be − 130 m from the present level. To simplify our model, we selected five variables from the 20 available when considering the analysis of variable contribution and response curves.
Results
Chloroplast non-coding regions
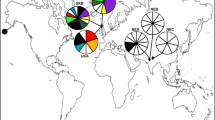
Variations from two chloroplast non-coding regions were investigated. We identified two haplotypes for Suaeda malacosperma with only two substitutions in the concatenated sequences (Online Resource 3) of trnH-psbA (359 bp) and rpl32-trnL (897 bp). The two substitutions were equal to the parsimonious informative site. The five representatives of each population shared only one of the two haplotypes. Haplotype A occurred in both Korea (GHW, SSA, BRY, SEC, and GYA) and Japan (SGA, HYM, YMG, and FKY), whereas haplotype B was found only in two populations (GJI and BSE) from the southern coast of Korea (Fig. 1a, Table 1). While haplotype A was widely distributed from the western Korean coast to Japan, haplotype B was restricted to the southern Korean coast.

Geographic distribution of cpDNA haplotypes and structure analysis in K = 3 of Suaeda malacosperma, and ΔK values in structure analysis. a Locality codes correspond to those in Table 1. Diameter of pie charts is associated with the number of samples. The circles surrounding pie charts correspond with the cpDNA haplotypes. Paleo-coast lines that were supposed to have formed during LGM were drawn based on ETOPO1 Global Relief Model. b ΔK values was estimated by using STRUCTURE HARVESTER (Earl and von Holdt 2012), according to the method of Evanno et al. (2005)
Genetic diversity
The number of nucleotide sites per population averaged 22,551.2, ranging from 18,796 for GYA to 25,518 for HYM (Table 1). Estimations of genetic diversity were based on a dataset of 120 loci. The mean values for nucleotide diversity (π) were 0.1350 for western Korea, 0.1186 for southern Korea, and 0.1460 for Japan. The lowest and highest values for π were 0.0866 (GJI) and 0.1713 (FKY). Observed heterozygosity and expected heterozygosity ranged from 0.1476 to 0.2198, and from 0.0845 to 0.1605, respectively. The percentage of polymorphic sites ranged from 0.1193 (GJI) to 0.2759 (BSE).
Population differentiation and genetic structure
Population differentiation (FST) average across all loci and populations was 0.2590, with pairwise population FST estimates ranging from 0.0085 between HYM and YMG in Japan, to 0.5185 between GJI and GYA on the southern coast of Korea (Table 2). The FST values among populations of western Korea (GHW, SSA, BRT, and SEC) and among Japanese populations (SGA, HYM, YMG, and FKY) were lower than the mean value. Furthermore, FST values (0.0085–0.1861) among populations in Japan were lower than those (0.1681–0.2311) among western Korean populations. Although populations along the southern Korean coast (GJI, BSE, and GYA) were geographically closer to Japanese populations than to western Korean populations, values for FST were larger among populations in Japan (0.2249–0.4421) than between Japanese and western Korean populations (0.1051–0.3682). Among all populations, GYA from southern Korea had the highest FST value.
Bayesian model-based clustering analysis of the MIG-Seq dataset for 120 loci revealed the highest ∆K at K = 3 (Fig. 1b). Three clusters were formed in the pie charts that summarized each run by using CLUMPAK (Fig. 1b): Cluster I (green; western coast of Korea, except for BRY), Cluster II (red; southern coast of Korea), and Cluster III (blue, Japan plus BRY from Korea). While most populations comprised a single ancestor lineage with little admixture, GYA in Cluster II was admixed with the ancestor lineage of Cluster III. Major genetic gaps occurred along the southwestern edge of the Korean Peninsula and the Korea/Tsushima Strait.
Distribution modeling
We conducted ENM of S. malacosperma based on altitude (alt) and four bioclimatic factors—mean temperature of the warmest quarter (bio10), precipitation in the driest month (bio14), precipitation in the wettest quarter (bio16), and precipitation in the warmest quarter (bio18). These environmental variables were selected to simplify our model when considering their individual contributions. The distribution prediction for current conditions (Fig. 2) reflected the current range of S. malacosperma (from the western coast to the southern coast of Korea, and from Kyushu to the southern coast of Honshu in Japan). As a measure of model fitness, the mean area under the receiver operating characteristic curve for testing data was high (0.988), which supported the predictive power of the model. The standard deviation was low (0.019) for the 10 replicates evaluated. The CCSM, MIROC, and MPI models for the LGM yielded different inferences for the paleo-distribution of S. malacosperma, despite the potential distribution being congruent in coastal regions from the continental shelf in the East China Sea to Kyushu through the southern coast of Korea.

Potential distributions during present time and LGM, inferred by distribution modeling
Discussion
Phylogeographic pattern and genetic diversity of Suaeda malacosperma
We used the MIG-Seq method and chloroplast non-coding regions to elucidate the phylogeographic pattern of the annual halophyte Suaeda malacosperma. Two putative genetic gaps emerged that separated three genetic clusters: western coast of Korea, southern coast of Korea, and the coastline of Japan. Results from our structure analysis with MIG-Seq were mostly concordant with the geographical pattern of the cpDNA haplotypes. However, there were some disagreements arose: two genetic discontinuities along the southwestern edge of the Korean Peninsula, and another in the Korea/Tsushima Strait. When the distribution of cpDNA haplotypes was examined, we found that although the first genetic discontinuity (i.e., the southwestern edge of the Korean Peninsula) was the same as that revealed in the structure analysis, the second one was detected between populations BSE and GYA on the southern Korean coast. This structure analysis further indicated that GYA has admixture of both Cluster II and Cluster III that are comprised of haplotype A, which appeared in populations from the western Korean coast and Japan. Since GYA has admixed lineages detected from structure analysis and haplotype B distributed in the western coast in Korea and Japan, we suggest that the Korea/Tsushima land bridge during the glacial period could have contributed to the gene flow from Japan to the southern coast of Korea. If one considers the rise in temperatures and sea level that accompanied a range shift toward the higher latitude, it is more likely from Japan via the Korea/Tsushima land bridge rather than from BRY on the western coast of Korea. As reported from previous research of land plants in East Asia (Chung et al. 2014; Lee et al. 2014; Jin et al. 2016), this repeated connection via the Korea/Tsushima land bridge during the Pleistocene (Kimura 1996; Ota 1998) might have acted as a temporary genetic corridor.
Although the genetic diversity of S. malacosperma was not correlated with variables of geographical distribution such as latitude, the Japanese populations had relatively low values for private alleles, and SSA and FKY revealed slightly higher genetic diversity (HE and π) when compared with the others. The distribution range for temperate plants has shifted due to climate change associated with oscillations in sea level during glacial periods. Those glacial periods have alternated several times with interglacial periods that featured rising temperatures. During those interglacial periods, range expansion occurred because suitable habitats extended farther north and into lower altitudes from refugia that had harbored genetic diversity during the glacial period. These processes shaped the general genetic patterns of many land plants, as reflected in “southern richness versus northern purity” through climatic fluctuation and oscillation in the Quaternary (Hewitt 1996, 2004). Several investigations of European coastal halophytes have revealed a consistency with the concept of “southern richness versus northern purity” (reviewed by Weising and Freitag 2007). However, the genetic pattern of S. malacosperma does not agree with that concept, possibly because of the loss of genetic diversity that resulted from significant range shifts after the glacial period. For example, potential habitats for this species in brackish areas where tides ebb and flow might have been affected by changes in locations, sediment deposits, and hydrology during those post-glacial range shifts. Those significant alterations may have influenced the patterns of genetic diversity for S. malacosperma.
Based on the output from our analyses of cpDNA and MIG-Seq data, the genetic structure of S. malacosperma does not seem to have been affected by sea currents associated with seed dispersal. Those currents are generally considered to be either contributors or barriers to the spread of coastal plants (Takayama et al. 2008; Westberg and Kadereit 2009; Han et al. 2015). In the Yellow Sea and the ECS around the Korean Peninsula and the western side of mainland Japan, circulation of sea currents shows seasonal fluctuations. Sudden season-related changes in currents have been detected in the Yellow Sea. For example, the Korean Coastal Current generally flows north counterclockwise in Summer but southward in Winter; the exception is the Cheju Warm Current, which is branched from the main current of the Kuroshio (Lie and Cho 2016). In contrast, the southern coast of Korea and the western side of Japan are influenced year-round by the Cheju Warm Current that flows eastward as well as by the northeastward-flowing Tsushima Warm Current and the southward-flowing Western Kyushu Current (Lie and Cho 2016). This discontinuity between the Korean Coastal Current and the Cheju Warm Current is in accord with the genetic gap at the southeastern edge of the Korean Peninsula that was revealed from our evaluation of genetic structure (Fig. 1a). Although this pattern may have resulted from the contribution of those currents as agents of dispersal, it cannot explain the genetic discontinuity between BRY and nearby populations or between BSE and GYA.
Large stretches of the western and the southern side of the Korean Peninsula show a complex ria-type coast that was formed during periods of glaciation (Castaing and Guilcher 1995). Because that type of coast harbors estuaries of rivers far from the sea, it is not or only weakly influenced by sea currents. Furthermore, the numerous islands on the western and southern edges of Korea can block those currents, such that the estuarial habitats of S. malacosperma are not affected. Based on the results from our analysis of genetic structure, we suspect that the populations of this species were isolated from each other because the ocean served as a geographical barrier. From their research on the estuarine Fucus ceranoides L. of northwestern Iberia, Neiva et al. (2012) have shown that its distinct phylogeographic structure has been shaped by density-barrier effects, but they do not dismiss the idea of oceanic dispersal. Instead, they have suggested that the effect of migration between established populations is too low to dilute their genetic differentiation that has been shaped through historical processes such as colonization. Even if dispersal of S. malacosperma by currents were possible, the migrants would not have been able to influence the gene frequency in already established populations.
Furthermore, the seed characters of S. malacosperma are not in favor of effective dispersal over longer distances. While several species within genus Suaeda produce heteromorphic seeds—lens-shaped black ones with a hard testa and pronounced dormancy, and disk-shaped brown ones with membranous testa and without dormancy (e.g., Cao et al. 2012; Yang et al. 2015)—S. malacosperma produces only olive-colored seeds of the latter type (Lee et al. 2007; Song et al. 2008; Wang et al. 2008; Yang et al. 2015). The absence of a protective seed coat and dormancy are the most likely factors associated with a short viability of the seeds.
Inference of past distribution and recolonization route based on ENM
The genetic structure of S. malacosperma was clustered into three geographic regions: western coast of Korea, southern coast of Korea, and Japan (Fig. 1a). Even though this species could migrate, gene flow was low among populations, which suggested that the recolonization routes and the possibility of genetically separated refugia varied for each region. The distribution model, predicted from five current variables, shows similarities between historic and contemporary pattern (Fig. 2). Furthermore, the coincidence among three paleo-distribution models inferenced different climate data that revealed a range in distribution patterns from the exposed continental shelf of the ECS to the main islands near the coast of Japan (Fig. 2). Based on the model for distribution during the LGM, S. malacosperma would have been found in estuarine environments similar to its present habitat. When transgression gradually occurred after the LGM, those types of environment shifted inland through paleo-river channels that were shaped in the Yellow Sea and on the southern edge of the Korean Peninsula (Yang et al. 2014; Yoo et al. 2016). This shifting of estuaries would have been accompanied by changes in habitat for S. malacosperma. When integrated with information about the genetic structure and the inference of past distribution and estuarine habitats of this species, we think it is more likely that recolonization within each of the three regions followed along the path of those estuaries as the sea level rose during the post-glacial period. In particular, it is plausible that populations on the western Korean coast, where the coastline was drastically altered during that period, were re-colonized from populations in the ECS shelf region that were distinct from those on the southern Korean coast. According to our results and inferences, the genetic structure of S. malacosperma distributed in the estuaries was determined by shifts in its historical range associated with climate change rather than by contemporary gene flow. This phenomenon has also been confirmed in coastal seaweeds that have an estuarine distribution (Neiva et al. 2012).
In our analysis of genetic structure and differentiation, BRY was genetically closer to populations in Japan than to those within the neighboring localities of the western coast of Korea. To explain that unexpected phenomenon we present two hypotheses: First, the BRY population was established from seed(s) of Japanese plants by accidental long-distance dispersal. Second, BRY might have originated from common ancestors that were distributed in the area from the ECS shelf to Japan, as deduced via ENM by past distribution during the LGM.
We have some basis for the first hypothesis. Waterfowl migration and maritime trade are considered means of dispersal that have contributed to the migration of plants from Europe to North America (Weising and Freitag 2007; Brandt et al. 2015). For example, some grebe and anatine species are residents or winter visitors to the coastal regions of Korea and Japan (Lee et al. 2005) while species such as Platalea minor are part of the East Asia/Australasia Flyway (Birdlife International 2018). For the second hypothesis, if populations along the western coast of Korea followed a similar route of recolonization from past distribution, it would be difficult to explain why the origin of BRY differed from that of others in the same region because gene flow among populations would have been frequent during the recolonization process. Therefore, considering the more recent establishment of plants following long-distance dispersal from Japan via waterfowl or maritime trade is more rational than assuming that the BRY population has remained relatively constant throughout the entire recolonization process.
Conservation of Suaeda malacosperma
Coastal regions are vulnerable to ongoing changes in climate (Nicholls and Cazenave 2010). Such unstable environments are influenced by sedimentation, water discharge, and altered patterns of water usage (Saito 2001). Habitats for the halophyte S. malacosperma are limited to brackish areas. The results from our cpDNA and MIG-Seq analyses showed that, due to their low capacity for dispersal, populations of this vulnerable species are isolated within various regions of western and southern Korea as well as along the coast of Japan. Our field survey also indicated that entire populations of S. malacosperma have been exposed to habitat fragmentation and reduction due to anthropogenic disturbances such as land reclamation (Suzuki 2003; Choi et al. 2014; Murray et al. 2014). Each of our sampling sites was located near dikes or artificial structures that present barriers between the sea and utilized land. Because of its estuarine pattern of distribution, the demographics for S. malacosperma populations at each locality are suffering from the impact of anthropogenic activities within those estuaries and tidal regions. They led to environmental changes in sedimentation, hydrology, and the extent of tidal areas that can have impacts on gene frequency within each population. In the past distribution inferred by ENM, the shift of range in Japan was lesser than other regions. Although it seems to be a condition that harbors more genetic diversity from the LGM than other populations, we were unable to identify significantly high genetic diversity in Japan as compared to other regions. The loss of genetic diversity could be influenced from not only significant range shifts after LGM, but also anthropogenic disturbances in the East Asian coastal region. Consequently, conservation strategies must be established to preserve genetic diversity and individual habitats. In doing so, one must also evaluate the three separate geographical clusters that define the genetic structure of that species. More diverse genetic components can be preserved if each region, coupled with each cluster, is treated as an independent conservation unit. According to our field survey, BRY on the western coast of Korea, BSE on the southern coast of Korea and YMG in Japan have large populations and relatively high genetic diversity than others. Faced with future global warming accompanied by changes in sea levels, the southern populations in Japan can be considered weaker than those in the north. Although the goal of conservation strategies needs to be focused on supporting the environment in which this species can survive, a detailed study of the ecological conditions in these estuarine environments is also important in order to establish suitable strategy.
References
Arafeh R, Kadereit JW (2006) Long-distance seed dispersal, clone longevity and lack of phylogeographical structure in the European distributional range of the coastal Calystegia soldanella (L.) R. Br. (Convolvulaceae). J Biogeogr 33:1461–1469. https://doi.org/10.1111/j.1365-2699.2006.01512.x
Birdlife International (2018) East Asia/Australasia flyway. Available at: http://datazone.birdlife.org/userfiles/file/sowb/flyways/8_EastAsia_Australasia_NEW.pdf. Accessed 31 Dec 2018
Brandt R, Lomonosova M, Weising K, Wagner N, Freitag H (2015) Phylogeny and biogeography of Suaeda subg. Brezia (Chenopodiaceae/Amaranthaceae) in the Americas. Pl Syst Evol 301:2351–2375. https://doi.org/10.1007/s00606-015-1233-y
Cao D, Baskin CC, Baskin JM, Yang F, Huang Z (2012) Comparison of germination and seed bank dynamics of dimorphic seeds of the cold desert halophyte Suaeda corniculata subsp. mongolica. Ann Bot (Oxford) 110:1545–1558. https://doi.org/10.1093/aob/mcs205
Carstens BC, Richards CL (2007) Integrating coalescent and ecological niche modeling in comparative phylogeography. Evolution (Lancaster) 61:1439–1454. https://doi.org/10.1111/j.1558-5646.2007.00117.x
Castaing P, Guilcher A (1995) Geomorphology and sedimentology of Rias. In: Perillo GME (ed) Developments in sedimentology, vol. 53. Elsevier, Oxford, pp 69–111
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Molec Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Choi YR (2014) Modernization, development and underdevelopment: reclamation of Korean tidal flats, 1950s–2000s. Ocean Coastal Managem 102:426–436. https://doi.org/10.1016/j.ocecoaman.2014.09.023
Choi SC, Choi DG, Hwang JS, Kim JG, Choo YS (2014) Solute patterns of four halophytic plant species at Suncheon Bay in Korea. J Ecol Environ 37:131–137. https://doi.org/10.5141/ecoenv.2014.016
Chung MY, López-Pujol J, Chung MG (2014) Genetic homogeneity between Korean and Japanese populations of the broad-leaved evergreen tree Machilus thunbergii (Lauraceae): a massive post-glacial immigration through the Korea Strait or something else? Biochem Syst Ecol 53:20–28. https://doi.org/10.1016/j.bse.2013.12.006
Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM (2009) The last glacial maximum. Science 325:710–714. https://doi.org/10.1126/science.1172873
Costanza R, d’Arge R, de Groot R, Farber S, Grasso M, Hannon B, Limburg K, Naeem S, O’Neill RV, Paruelo J, Raskin RG, Sutton P, van den Belt M (1997) The value of the world’s ecosystem services and natural capital. Nature 387:253–260. https://doi.org/10.1038/387253a0
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resources 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Escudero M, Vargas P, Arens P, Ouborg NJ, Luceño M (2010) The east-west-north colonization history of the Mediterranean and Europe by the coastal plant Carex extensa (Cyperaceae). Molec Ecol 19:352–370. https://doi.org/10.1111/j.1365-294X.2009.04449.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molec Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
García-Fernández A, Vitales D, Pellicer J, Garnatje T, Vallès J (2017) Phylogeographic insights into Artemisia crithmifolia (Asteraceae) reveal several areas of the Iberian Atlantic coast as refugia for genetic diversity. Pl Syst Evol 303:509–519. https://doi.org/10.1007/s00606-017-1387-x
Gent PR, Danabasoglu G, Donner LJ, Holland MM, Hunke EC, Jayne SR, Lawrence DM, Neale RB, Rasch PJ, Vertenstein M, Worley PH, Yang ZL, Zhang M (2011) The community climate system model version 4. J Clim 24:4973–4991. https://doi.org/10.1175/2011JCLI4083.1
Han Q, Higashi H, Mitsui Y, Setoguchi H (2015) Distinct phylogeographic structures of wild radish (Raphanus sativus L. var. raphanistroides Makino) in Japan. PLoS ONE 10:e0135132. https://doi.org/10.1371/journal.pone.0135132
Hara H (1942) Hirohamatsuna. J Jap Bot 18:26–28 (in Japanese)
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. https://doi.org/10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans Roy Soc London 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Jakob SS, Ihlow A, Blattner FR (2007) Combined ecological niche modelling and molecular phylogeography revealed the evolutionary history of Hordeum marinum (Poaceae)—niche differentiation, loss of genetic diversity, and speciation in Mediterranean Quaternary refugia. Molec Ecol 16:1713–1727. https://doi.org/10.1111/j.1365-294X.2007.03228.x
Jin DP, Lee JH, Xu B, Choi BH (2016) Phylogeography of East Asian Lespedeza buergeri (Fabaceae) based on chloroplast and nuclear ribosomal DNA sequence variations. J Pl Res 129:793–805. https://doi.org/10.1007/s10265-016-0831-2
Kadereit JW, Westberg E (2007) Determinants of phylogeographic structure: a comparative study of seven coastal flowering plant species across their European range. Watsonia 26:229–238
Kadereit JW, Arafeh R, Somogyi G, Westberg E (2005) Terrestrial growth and marine dispersal? Comparative phylogeography of five coastal plant species at a European scale. Taxon 54:861–876. https://doi.org/10.2307/25065473
Kimura M (1996) Quaternary paleogeography of the Ryukyu Arc. J Geogr (Tokyo) 105:259–285. https://doi.org/10.5026/jgeography.105.3_259 (in Japanese)
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Molec Ecol Resources 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Lassmann T, Hayashizaki Y, Daub CO (2009) TagDust: a program to eliminate artifacts from next generation sequencing data. Bioinformatics 25:2839–2840. https://doi.org/10.1093/bioinformatics/btp527
Lee WS, Koo TH, Park JY (2005) A field guide to the birds of Korea, 2nd edn. LG Evergreen Foundation, Seoul
Lee JS, Park DS, Ihm BS, Lee WJ (2007) Taxonomic reappraisal on Suaeda australis (Chenopodiaceae) in Korea based on the morphological and molecular characteristics. J Pl Biol 50:605–614. https://doi.org/10.1007/BF03030603
Lee JH, Lee DH, Choi BH (2013) Phylogeography and genetic diversity of East Asian Neolitsea sericea (Lauraceae) based on variations in chloroplast DNA sequences. J Pl Res 126:193–202. https://doi.org/10.1007/s10265-012-0519-1
Lee JH, Lee DH, Choi IS, Choi BH (2014) Genetic diversity and historical migration patterns of an endemic evergreen oak, Quercus acuta, across Korea and Japan, inferred from nuclear microsatellites. Pl Syst Evol 300:1913–1923. https://doi.org/10.1007/s00606-014-1017-9
Lee DH, Lee JH, Cho WB, Choi BH (2016) The establishment history of alpine Leontopodium japonicum (Asteraceae) resembles that of warm-temperate plants on the Korean Peninsula. Pl Syst Evol 302:1483–1494. https://doi.org/10.1007/s00606-016-1346-y
Lie HJ, Cho CH (2016) Seasonal circulation patterns of the Yellow and East China Seas derived from satellite-tracked drifter trajectories and hydrographic observations. Progr Oceanogr 146:121–141. https://doi.org/10.1016/j.pocean.2016.06.004
Merow C, Smith MJ, Silander JA Jr (2013) A practical guide to MaxEnt for modeling species’ distributions: what it does, and why inputs and settings matter. Ecography 36:1058–1069. https://doi.org/10.1111/j.1600-0587.2013.07872.x
Murray NJ, Clemens RS, Phinn SR, Possingham HP, Fuller RA (2014) Tracking the rapid loss of tidal wetlands in the Yellow Sea. Frontier Ecol Environm 12:267–272. https://doi.org/10.1890/130260
Nakanishi H (2001) Phytosociological study of a Suaeda malacosperma community and distribution of Suaeda species in western Kyushu. Veg Sci 18:99–106. https://doi.org/10.15031/vegsci.18.99 (in Japanese)
National Institute of Biological Resources (2012) Korean red list of threatened species: mammalis, birds, reptiles, amphibians, fishes and vascular plants. National Institute of Biological Resources, Incheon
Neiva J, Pearson GA, Valero M, Serrão EA (2012) Fine-scale genetic breaks driven by historical range dynamics and ongoing density-barrier effects in the estuarine seaweed Fucus ceranoides L. BMC Evol Biol 12:78. https://doi.org/10.1186/1471-2148-12-78
Nicholls RJ, Cazenave A (2010) Sea-level rise and its impact on coastal zones. Science 328:1517–1520. https://doi.org/10.1126/science.1185782
Ota H (1998) Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu archipelago, Japan, with special reference to their paleogeographical implications. Res Populat Ecol 40:189–204. https://doi.org/10.1007/Bf02763404
Park JS, Choi IS, Lee DH, Choi BH (2018) The complete plastid genome of Suaeda malacosperma (Amaranthaceae/Chenopodiaceae), a vulnerable halophyte in coastal regions of Korea and Japan. Mitochondrial DNA B Resources 3:382–383. https://doi.org/10.1080/23802359.2018.1437822
Prinz K, Weising K, Hensen I (2013) Habitat fragmentation and recent bottlenecks influence genetic diversity and differentiation of the central European halophyte Suaeda maritima (Chenopodiaceae). Amer J Bot 100:2210–2218. https://doi.org/10.3732/ajb.1300097
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qi X-S, Chen C, Comes HP, Sakaguchi S, Liu YH, Tanaka N, Sakio H, Qiu YX (2012) Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytol 196:617–630. https://doi.org/10.1111/j.1469-8137.2012.04242.x
Qiu YX, Fu CX, Comes HP (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Molec Phylogen Evol 59:225–244. https://doi.org/10.1016/j.ympev.2011.01.012
Ryu JS, Choi JW, Kang SG, Koh CH, Huh SH (1997) Temporal and spatial changes in the species composition and abundance of benthic polychaetes after the construction of Shihwa dike (west coast of Korea). J Korean Soc Oceanogr 2:101–109 (in Korean)
Saito Y (2001) Deltas in southeast and East Asia: their evolution and current problems. In: Mimura N, Yokoki H (eds) Proceedings of APN/SURVAS/LOICZ joint conference on coastal impacts of climate change and adaptation in the Asia-Pacific region. Kobe, Japan, pp 185–191
Sang T, Crawford DJ, Stuessy TF (1997) Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Amer J Bot 84:1120–1136. https://doi.org/10.2307/2446155
Sato S (2006) Drastic change of bivalves and gastropods caused by the huge reclamation projects in Japan and Korea. Plankt Benthos Res 1:123–137. https://doi.org/10.3800/pbr.1.123
Shaw J, Lickey EB, Schilling EE, Small RL (2007) Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Amer J Bot 94:275–288. https://doi.org/10.3732/ajb.94.3.275
Shim HB, Chung JY, Choi BH (2001) One unrecorded species from Korea: Suaeda malacosperma Hara. Korean J Pl Taxon 31:383–387. https://doi.org/10.11110/kjpt.2001.31.4.383 (in Korean)
Shim HB, Cho WB, Choi BH (2009) Distribution of halophytes in coastal salt marsh and on sand dunes in Korea. Korean J Pl Taxon 39:264–276. https://doi.org/10.11110/kjpt.2009.39.4.264 (in Korean)
Song J, Fan H, Zhao Y, Jia Y, Du X, Wang B (2008) Effect of salinity on germination, seedling emergence, seedling growth and ion accumulation of a euhalophyte Suaeda salsa in an intertidal zone and on saline inland. Aquatic Bot 88:331–337. https://doi.org/10.1016/j.aquabot.2007.11.004
Suyama Y, Matsuki Y (2015) MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci Rep 5:16963. https://doi.org/10.1038/srep16963
Suzuki T (2003) Economic and geographic backgrounds of land reclamation in Japanese ports. Mar Pollut Bull 47:226–229. https://doi.org/10.1016/S0025-326X(02)00405-8
Takahashi Y, Suyama Y, Matsuki Y, Funayama R, Nakayama K, Kawata M (2016) Lack of genetic variation prevents adaptation at the geographic range margin in a damselfly. Molec Ecol 25:4450–4460. https://doi.org/10.1111/mec.13782
Takayama K, Tateishi Y, Murata J, Kajita T (2008) Gene flow and population subdivision in a pantropical plant with sea-drifted seeds Hibiscus tiliaceus and its allied species: evidence from microsatellite analyses. Molec Ecol 17:2730–2742. https://doi.org/10.1111/j.1365-294X.2008.03799.x
Tamaki I, Yoichi W, Matsuki Y, Suyama Y, Mizuno M (2017) Inconsistency between morphological traits and ancestry of individuals in the hybrid zone between two Rhododendron japonoheptamerum varieties revealed by a genotyping-by-sequencing approach. Tree Genet Genomes 13:4. https://doi.org/10.1007/s11295-016-1084-x
UNEP (2006) Marine and coastal ecosystems and human well-being: a synthesis report based on the findings of the Millennium Ecosystem Assessment. UNEP, Nairobi
Wang L, Huang Z, Baskin CC, Baskin JM, Dong M (2008) Germination of dimorphic seeds of the desert annual halophyte Suaeda aralocaspica (Chenopodiaceae), a C4 plant without Kranz Anatomy. Ann Bot (Oxford) 102:757–769. https://doi.org/10.1093/aob/mcn158
Watanabe S, Hajima T, Sudo K, Nagashima T, Takemura T, Okajima H, Nozawa T, Kawase H, Abe M, Yokohata T, Ise T, Sato H, Kato E, Takata K, Emori S, Kawamiya M (2011) MIROC-ESM 2010: model description and basic results of CMIP5-20c3m experiments. Geosci Model Developm 4:845–872. https://doi.org/10.5194/gmd-4-845-2011
Weising K, Freitag H (2007) Phylogeography of halophytes from European coastal and inland habitats. Zool Anz 246:279–292. https://doi.org/10.1016/j.jcz.2007.07.005
Westberg E, Kadereit JW (2009) The influence of sea currents, past disruption of gene flow and species biology on the phylogeographical structure of coastal flowering plants. J Biogeogr 36:1398–1410. https://doi.org/10.1111/j.1365-2699.2008.01973.x
Yang S, Wang Z, Dou Y, Shi X (2014) A review of sedimentation since the Last Glacial Maximum on the continental shelf of eastern China. Geol Soc London Mem 41:293–303. https://doi.org/10.1144/M41.21
Yang F, Baskin JM, Baskin CC, Yang X, Cao D, Huang Z (2015) Effects of germination time on seed morph ratio in a seed-dimorphic species and possible ecological significance. Ann Bot (Oxford) 115:137–145. https://doi.org/10.1093/aob/mcu210
Yoichi W, Kawamata I, Matsuki Y, Suyama Y, Uehara K, Ito M (2018) Phylogeographic analysis suggests two origins for the riparian azalea Rhododendron indicum (L.) Sweet. Heredity 121:594–604. https://doi.org/10.1038/s41437-018-0064-3
Yoo DG, Lee GS, Kim GY, Kang NK, Yi BY, Kim YJ, Chun JH, Kong GS (2016) Seismic stratigraphy and depositional history of late Quaternary deposits in a tide-dominated setting: an example from the eastern Yellow Sea. Mar Petrol Geol 73:212–227. https://doi.org/10.1016/j.marpetgeo.2016.03.005
Acknowledgements
We thank J. Ota, H. Tsubota, and H. Nakanishi at Hiroshima University for help in collecting samples in Japan. We are also grateful to our colleagues I. S. Choi, D. P. Jin, and J. W. Park at the Plant Systematics Laboratory of Inha University for assistance in collecting samples and in discussing the manuscript. This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2015R1D1A1A01059886).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The authors comply with all rules of the journal following the COPE guidelines; all authors have contributed and approved the final manuscript.
Additional information
Handling Editor: Christian Parisod.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. Occurrence data of Suaeda malacosperma used in Maxent program.
Online Resource 2. The list of 19 bioclimatic data used in Maxent program.
Online Resource 3. Polymorphic sites and chloroplast DNA haplotypes based on sequences of trnH-psbA and rpl32-trnL regions from Suaeda malacosperma.
Rights and permissions
About this article

Cite this article
Park, JS., Takayama, K., Suyama, Y. et al. Distinct phylogeographic structure of the halophyte Suaeda malacosperma (Chenopodiaceae/Amaranthaceae), endemic to Korea–Japan region, influenced by historical range shift dynamics. Plant Syst Evol 305, 193–203 (2019). https://doi.org/10.1007/s00606-018-1562-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-018-1562-8