
Abstract
Neolitsea sericea is an evergreen broad leaved tree in the warm-temperate regions of East Asia. This area is a hotspot for plant species richness and endemism caused by dynamic changes in land configuration during the Quaternary. However, the historical migration of such evergreen tree species is still poorly understood. In an attempt to reconstruct the phylogeographic history of N. sericea during the Quaternary, we identified the chloroplast DNA haplotypes of 287 individuals from 33 populations covering almost all of its geographic range. Analyses were based on sequence data from the trnL–F, psbC–trnS, and rps16 regions. Nine haplotypes were identified. The majority included ancestral types in the southwestern part of the main islands of Japan, with other region-specific haplotypes being found in populations on the Korean Peninsula, Taiwan (Isl. Lanyu), and elsewhere in Japan. A statistical parsimony network revealed two lineages derived from Japanese main islands. One was represented on the Korean Peninsula, the other on Isl. Lanyu. The current distribution of N. sericea has been shaped by colonization via land bridges. During the glacial periods, two primary, but separate migration routes were followed—from the southwestern part of the Japanese main islands to either the Korean Peninsula or Taiwan. In addition, we believe the Zhoushan populations were shaped by post-glacial processes through an ECS land bridge (East China Sea basin) from northern refugia that existed during the late Pleistocene.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Global climatic oscillations during the Quaternary have had a strong influence on species distribution and evolution. Thus, the present patterns of plant and animal species are the result of migration and extinction due to climatic changes (Hewitt 2000, 2003). In East Asia, fluctuations in sea levels that accompanied those oscillations produced dynamic changes in land configurations during the Quaternary (Kimura 1996, 2000; Kizaki and Oshiro 1977; Ota 1998). They have affected the fragmentation and rejoining of habitats for temperate plant species (Harrison et al. 2001; Qian and Ricklefs 2000). The modern genetic structure for temperate plant taxa distributed in that region would have been formed through expansion and contraction via land bridges (e.g., Bai et al. 2010; Li et al. 2008; Qiu et al. 2009a, b). Those warm-temperate climate zones of East China, southern Japan, and the southern tip of Korea are now hotspots for plant species richness and endemism (Qian and Ricklefs 2000; Xie 1997).
A simulated paleo-vegetation reconstruction by Harrison et al. (2001) has suggested that temperate forests are limited to a narrow belt at low elevations across the continental shelf that linked China, Korea, and Japan during the Last Glacial Maximum (LGM). In particular, warm-temperate evergreen forests that belong to temperate forest biomes are more extremely restricted than are temperate deciduous forests. However, this basic Quaternary expansion–contraction (EC) model of latitudinal range change is not paradigmatic due to differences in life cycles, habitat ecology, and a species’ ability to disperse (e.g., Hewitt 1999; Qiu et al. 2011; Taberlet et al. 1998). Several studies in East Asia have led researchers to formulate various opinions about the historical migration of temperate taxa (Bai et al. 2010; Chen et al. 2008; Qiu et al. 2009b). However, such migration by warm-temperate evergreen tree species in this region is poorly understood. Few phylogeographical examinations have been made of East Asian species, and most population genetics studies have focused on very localized areas, such as China, Korea, Japan, or Taiwan (Aoki et al. 2006; Cheng et al. 2005; Chung et al. 2000; Wang et al. 2005).
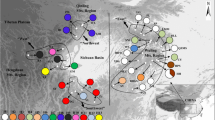
Neolitsea sericea (Blume) Koidz. (Lauraceae) is an evergreen broad leaved tree growing in East Asian warm-temperate regions. It is commonly found in the evergreen forests of Korea and Japan, and shows a disjunctive and restricted distribution on Lanyu Island of Taiwan and Zhoushan Archipelago of East China (Fig. 1) (Lee and Choi 2010). The Korean Peninsula and Japanese main islands represent the northwestern- and northeastern-most limits of its range (Ohashi et al. 2006; Lee and Choi 2010). This species is represented by two varieties: var. sericea and var. aurata. The latter is distinguished by its persistent golden-brown hairs on the lower surfaces of the leaves (Hatusima 1969). Its habitats are restricted to Lanyu and Ryukyu (Chen and Pan 1997; Hatusima 1969; Liao 1996). As such, it is a suitable model for obtaining historical information as it relates to climatic changes in the Quaternary.

Geographic distribution of cpDNA haplotypes detected in Neolitsea sericea. Dark shading indicates current mainland and island configurations; light shading shows exposed coastal areas and sea basins of East Asia during the time of glacial-induced alterations in sea levels at late Pleistocene. Suppositions are based on a hypothesis by Kizaki and Oshiro (1977), then modified according to that of Hikida and Ota (1997)
However, Zhai et al. (2012) have suggested that N. sericea has two distinct lineages in areas north and south of the ‘Tokara Gap’, i.e., a sea strait between northern and central Ryukyu. Their findings serve to increase our understanding of allopatric speciation in East Asian plant endemics (Zhai et al. 2012). Nevertheless, we still lack a clear picture of the phylogeographic structure for this species, especially within the Korean Peninsula. That region has played an important evolutionary role, and could have provided conditions suitable for generating new species via allopatric speciation, through the contemporary fragmentation of temperate forest combined with fluctuations in climate and sea levels (see Qiu et al. 2009b). Therefore, by analyzing that genetic structure on the Korean Peninsula, in essence covering its entire range, we can correctly acquire phylogeographical information for this species.
Our understanding of the processes that have influenced current distributions of populations and species has been greatly enhanced by the concept of phylogeography. This approach can be utilized to infer historical scenarios and account for spatial arrangements (Avise 2000). Chloroplast (cp) DNA is non-recombinant and maternally inherited (Corriveau and Coleman 1988), and it displays slow sequence evolution (Wolfe et al. 1987). Thus, cpDNA markers are useful tools for phylogeographic evaluations (Aoki et al. 2004, 2006; Heuertz et al. 2004; Petit et al. 2002; Rendell and Ennos 2003; Sugahara et al. 2011).
As has been observed with other temperate species in East Asia (Qiu et al. 2009b), we might expect that the distribution and genetic structure of N. sericea are deeply linked to paleogeography on the Korean Peninsula during the Quaternary (<2 million years ago). Therefore, to reconstruct its phylogeographic history, we investigated its variations in cpDNA sequences, paying particular attention to Korean populations. Our specific goals were to: (1) elucidate the genetic diversity within and among populations based on those sequences, (2) infer its historical migration routes in East Asia during the Quaternary, and (3) examine the taxonomic entity of N. sericea var. aurata.
Materials and methods
Sampling
Plants of Neolitsea sericea var. sericea were sampled from 32 populations in Korea, Japan, and China (Isl. Putuo of Zhoushan Archipelago). One population of var. aurata was chosen from Taiwan (Isl. Lanyu). These populations represented almost the entire distributional range of this species except for the Islands of Ryukyu and Ogasawara. From each population, 3 to 13 plants were taken, for a total of 287 individuals (Table 1). Voucher specimens for these samples were deposited in the Herbarium at Inha University (IUI) (Table S1).
DNA extraction and PCR amplification
Genomic DNA was extracted from fresh leaves, using a G-spin™ IIp Kit for plants (iNtRON, Seongnam, Korea). After preliminary screening of eight non-coding regions for cpDNA (trnL–F, rps16, rpl16, psbA–trnH, psbC–trnS, rpoB–trnC, atpB–rbcL, and petD–rpoA), we selected trnL–F, psbC–trnS, and rps16 for the full survey because most of the variation was observed in those three regions (Lee et al. 2011). The regions were amplified by polymerase chain reaction (PCR). Primers c and f (Taberlet et al. 1991) were used to amplify the trnL intron and the trnL–trnF intergenic spacer (trnL–F). Primers psbC–trnS/F, psbC–trnS/R, rps16/2F, and rps16/2R (Nishizawa and Watano 2000) were used to amplify the non-coding regions of the psbC–trnS intergenic spacer and rps16 intron.
PCR was conducted with a GeneAmp® PCR System 2700 Thermal Cycler (Applied Biosystems, Foster City, CA, USA). Each reaction mixture contained 200 μM dNTPs (GeneCraft, Lüdinghausen, Germany), 1× PCR buffer with 1.5 mM MgCl2, 1 U of Taq DNA polymerase (TaKaRa, Seoul, Korea), 10 ng of DNA, and an appropriate concentration of primers in a total volume of 50 μL. Conditions included an initial denaturation at 94 °C for 2 min; followed by 35 cycles at 94 °C for 30 s, 52 °C for 45 s, and 72 °C for 1 min; with a final extension at 72 °C for 10 min. PCR products were visualized on 2 % agarose gels, purified with a high Pure PCR product Purification Kit (iNtRON), and sequenced with an ABI 3100 genetic Analyzer, using the ABI BigDye™ Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). These sequences were deposited with their accession numbers in the GenBank database (Table S1).
Data analysis
All sequences were aligned manually with the program Clustal X version 1.83 (Thompson et al. 1997). Each indel was treated as a one-point mutation. The cpDNA haplotypes were determined based on those aligned sequences. The genealogical degree of relatedness among haplotypes was represented by a statistical parsimony network, generated by the program TCS version 1.21 (Clement et al. 2000). In addition, N. aciculata and N. villosa served as the outgroup to determine the ancestral haplotype.
Chloroplast DNA haplotype diversity (Nei 1987) was calculated for each population (h S) and for the overall range (h T), using DnaSP version 3.53 (Rozas and Rozas 1999). Two coefficients for gene differentiation—G ST (all populations) and N ST (differentiation influenced by both haplotype frequencies and genetic distances between haplotypes)—were estimated from cpDNA haplotypes following the methods of Pons and Petit (1996) and using the program PERMUT (available at http://www.Pierroton.inra.fr/genetics/labo/Software/Permut/). To test for the presence of a phylogeographical structure, we compared values for G ST and N ST with a permutation test that used 10,000 permutations. If N ST > G ST, then the closely related haplotypes occurred in the same populations, thereby indicating that such a structure existed (Pons and Petit 1996).
Population differentiation for between-region comparisons was assessed by non-hierarchical analysis of molecular variance (AMOVA; Excoffier et al. 1992), using the program ARLEQUIN version 3.0 (Excoffier et al. 2005). We did not choose a suitable hierarchical model for calculating among-region differentiation via ΦCT because this would have required multiple populations and only one population each exists in East China and Taiwan.
Mantel tests (Mantel 1967) were performed to test the correlation between the matrix of pair-wise ΦST values and the matrix of geographical distances between pairs of populations using ARLEQUIN version 3.0; AMOVA and Mantel tests were used to evaluate significance with 10,000 permutations.
The possibility of a geographical structure was also investigated by spatial analysis of molecular variance, using the program SAMOVA version 1.0 (Dupanloup et al. 2002). This method is based on a simulated annealing procedure that defines groups of populations that are geographically adjacent, geographically homogeneous, and maximally differentiated from each other. The most likely number of groups (K) was identified by repeatedly running the program for 10,000 iterations for K ∈ {2,…,15}, using 100 random initial conditions, then retaining the largest ΦCT values (i.e., the greatest proportion of total genetic variance due to differences between groups) as predictors of the best grouping of populations (Dupanloup et al. 2002).
Results
Variation and distribution of haplotypes
We sequenced three cpDNA regions from 287 individuals (33 populations) of Neolitsea sericea. Among these, the trnL–F (868–925 bp) region was the most variable, having three polymorphisms, i.e., a polynucleotide repeat-length and insertion-deletion (indel) that included a 52-bp mutation as well as a nucleotide substitution. The rps16 (261–265 bp) region had two polymorphisms, including a nucleotide substitution and mononucleotide repeat-length, while the psbC–trnS (241 bp) region had only one polymorphism, a nucleotide substitution. When combined, their sequences were aligned for a consensus length of 1,431 bp. Based on these six polymorphisms, nine haplotypes were recognized for N. sericea (Table 2).
The distribution and frequencies of haplotypes A to I among the 33 sampled populations of var. sericea and var. aurata are shown in Fig. 1 and Table 1. No dominant haplotype was widespread over the entire distributional range of this species. Haplotype E occurred at high frequency in the populations from the Korean Peninsula and East China. Haplotypes A, C, and D were found at high frequency in Japan, with haplotype A occurring only in that country. Haplotype G was distributed in two disjunctive areas—the Chugoku region of Japan and Isl. Lanyu of Taiwan. In addition, three region-specific haplotypes (F, H, and I) were found in a few populations: F from populations 7 and 13 of the Korean Peninsula; H from population 33 of Isl. Lanyu; and I from populations 27 and 30 of Japan. Among the nine haplotypes identified in this species, all but two (F and H) were in Japan. Ten populations comprised just a single haplotype while the other 23 had two or more (Fig. 1).
Our SAMOVA results revealed that values for F CT decreased progressively as the number of groups increased, and no geographical grouping of populations was apparent. In fact, each new group that was delimited was represented by a single population or else two geographically distant populations (data not shown).
Regional differences in haplotype diversity
Total genetic diversity (h T = 0.713), based on cpDNA data across all populations, was much higher than the average within-population diversity (h s = 0.390). A phylogeographic structure was found over the entire range and in Japan, but neither N ST value was significantly higher than its corresponding G ST value (range: N ST = 0.485 > G ST = 0.458; Japan: N ST = 0.263 > G ST = 0.260). By contrast, no such structure was found on the Korean Peninsula (G ST = 0.634 > N ST = 0.616). At the regional level, the highest average within-population diversity was detected in Japan (h s = 0.592); East China (Isl. Putuo) and Taiwan (Isl. Lanyu) had values higher than the average whereas the Korean Peninsula had the lowest (below-average) value (h s = 0.092; Table 1).
When compared among all regions, estimated divergences in terms of ΦST revealed that the Korean Peninsula, Isl. Putuo, and Isl. Lanyu were more genetically similar to Japan than to other regions. In each instance, highly significant genetic differentiation was found between Taiwan (Isl. Lanyu) and the Asiatic continent (Korean Peninsula: ΦST = 0.990, East China: ΦST = 0.960; Table 3). The Mantel tests showed no significant correlation between population differentiation and geographical distance within either region (Korean Peninsula: r = −0.044, P < 0.547 vs. Japan: r = 0.020, P < 0.356). However, a significant positive correlation was noted over the entire range (r = 0.428, P < 0.001).
Phylogenetic relationships of haplotypes
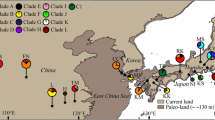
Phylogenetic relationships among haplotypes were revealed in the statistical parsimony network (Fig. 2). All haplotypes were distinguished from each other by one or two mutational steps. Statistical parsimony analysis at the 95 % confidence limit initially resolved two separate networks—one including haplotypes G, H, and I, versus the remaining haplotypes. This was due to a 52-bp indel between the two groups, which could be joined only with a non-parsimonious connection (Fig. 2).

Relatedness among chloroplast haplotypes (A–I) detected in Neolitsea sericea, represented in a statistical parsimony network. Identities of haplotypes are described in Table 2. Two phylogeographic lineages, derived from putative basal groups A, B, C, and D, are indicated by circles in black (Lineage Ι: E and F) and grey (Lineage ΙΙ: G, H, and I)
Haplotypes A and/or B were supported as basal types. The highest root probability was assigned by TCS as haplotype B (P = 0.260). In our outgroup comparison, the closely related N. villosa and N. aciculata had haplotypes A and B (data not shown). Furthermore, those two were located at the center of the network (Fig. 2). By contrast, haplotype C was distinguished from haplotypes A and B only by differences in their mononucleotide repeat-lengths. Thus, we assumed that haplotypes A, B, C, and D would be the basal group of N. sericea. In addition, two groups derived from the basal group were found in our statistical parsimony network—haplotypes G, H, and I, versus haplotypes E and F.
Discussion
Genetic diversity and population structure
Our cpDNA analysis of 33 East Asian populations of Neolitsea sericea revealed a high degree of total haplotype diversity (h T = 0.713). This is generally thought to reflect a long evolutionary history (Huang et al. 2001). The average within-population diversity (h s = 0.392) was considerably lower than the total haplotype diversity, probably because of the southwestern coastal populations (1–11) on the Korean Peninsula. Many of them (1–6, 9–11) occur in fragmented or small habitats that comprised a single haplotype, except for Isl. Putuo of East China. By contrast, populations found in continuous or large habitats had two or more haplotypes. This result is consistent with the theoretical expectation that larger populations have greater genetic diversity in the absence of selection (Kimura 1983).
Plants of N. sericea have an endozoochory type of seed dispersal that relies upon ingestion by vertebrate animals, such as monkeys and birds (Hong et al. 2007; Kominami et al. 2003). Species that produce ingestible seeds may exhibit weak population differentiation (e.g., Petit et al. 2003). However, the only life-history trait for maternally inherited markers that has been found to be significantly correlated with genetic differentiation among populations is the mode of seed dispersal (gravity vs. other means) (Duminil et al. 2007). We noted significant genetic divergence between the geographically adjacent Korean Peninsula and Japanese main islands (ΦST = 0.513; Table 3). This also indicated that genetic exchange between regions through the action of birds was not frequent over long periods. In fleshy-fruited European Frangula alnus and Japanese Photinia glabra, high genetic differentiation and a clear phylogeographical structure have been detected (Aoki et al. 2006; Hampe et al. 2003). The pattern of population differentiation identified here in N. sericea is not random, and genealogy is associated with the geographical distribution of cpDNA haplotypes.
Here, we identified seven of the nine haplotypes in the southwestern portion of those main islands (i.e., Kyushu, Shikoku, and Chugoku). Such haplotype richness provides a strong possibility for the existence of primary and persistent refugia in that area. During post-glacial re-colonization, the ancestral haplotypes were widespread, making it difficult to trace the sites for those glacial refugia, a process that often relies on derived or unique haplotypes for purposes of identification (Cheng et al. 2005). In fact, populations on the Korean Peninsula and in Japan and Taiwan (Isl. Lanyu) did have region-specific haplotypes (F, I, and H, respectively), which suggests that those locations were three separate refugia.
Our results indicated two geographically different haplotype lineages—I (E and F) and II (G, H, and I). The putative basal group (A, B, C, and D) showed a higher frequency in Japan than in other regions, and ancestral haplotypes A and B were distributed only in that country. By contrast, lineages I and II derived from the ancestral group were distributed in each of the other regions—I, on the Korean Peninsula, and II, in Taiwan (Isl. Lanyu). For comparison, East China (Isl. Putuo) did have haplotype C for the basal group and haplotype E within Lineage I.
Consequently, we suggest that population structuring of this species was shaped by colonization via land bridges. In particular, two main, but separate, migration routes were run during the glacial periods—from the southwestern part of the Japanese main islands to either Korean Peninsula or Taiwan.
Historical phylogeography and interpreting the genetic structure between Korea and Japan populations
During the glacial periods, the main islands of Japan (Kyushu, Shikoku, Honshu, and Hokkaido) were connected to the Asiatic continent (Korean Peninsula and East China) several times via paleogeographic land bridges (e.g., the Korean/Tsushima Strait, the East China Sea basin, and the Ryukyu Islands) (Kimura 1996, 2000; Kitamura and Kimoto 2006; Kizaki and Oshiro 1977; Ota 1998). In Korea and Japan, over the interval from 0 to 1.71 million years ago, the Korean/Tsushima Current Sea flowed into the East Sea (the Sea of Japan) during interglacial stages (Kitamura and Kimoto 2006). These periodical openings of the Korean/Tsushima Strait would have acted as a barrier to persistent contact of Neolitsea sericea through a land bridge between the Korean Peninsula and the Japanese main islands. In contrast, the periodic formation of a land bridge would have countervailed any genetic differentiation of N. sericea populations between the Korean Peninsula and those Japanese islands. Thus, the population structure of these two regions would have been modulated by a balance between vicariance and immigration through historical processes that were linked to the Quaternary paleogeography. The phylogeographical structure of Korean populations is thought to have been derived from a small Korean glacial refugium, plus re-colonization from eastern Japanese refugia.
During the glacial periods, East China, like the Korean Peninsula, was repeatedly connected with the Japanese main islands by a land bridge over the ECS basin (Kimura 1996, 2000). However, in the early and middle Pleistocene, East China and the southern part of the Korean Peninsula would have been separated by the East China Sea (Kizaki and Oshiro 1977; Ota 1998; Fig. S1). This wide oceanic barrier would apparently have limited migration into East China from the Korean Peninsula and Japan. Moreover, wide stretches along the continental shelf of the East China Sea were exposed during the late Pleistocene (Kizaki and Oshiro 1977; Ota 1998). This might have provided ample opportunities for range expansion into East China. In addition, because we found no unique haplotypes at Isl. Putuo, we propose that, compared with the Korean populations, the disjunctive distribution of the Putuo population is relatively recent, possibly being post-glacial rather than evolving allopatrically due to vicariance during the early or middle Pleistocene. Our conclusion is consistent with that of Zhai et al. (2012), who suggested that species within the Zhoushan populations are remnants of a lineage that extended along the exposed ECS basin during the LGM, and that they colonized the area before the island became isolated (c. 9000 BP–7000 BP; Yiming et al. 1998).
By comparison, during the late Pleistocene, a connection is evident between the Asiatic continent and the Japanese main islands while a link between Japan and Taiwan is very weak (Kizaki and Oshiro 1977; Ota 1998; Fig. 1). Thus, our results show that much greater genetic divergence occurred between Japan and Taiwan than between Japan and the Asiatic continent, and that a completely different lineage of the haplotype network exists between the Asiatic continent and Taiwan. To explain this, we might assume that the Tokara Gap between central and northern Ryukyu acted as a strong barrier, causing the genetic lineages of N. sericea to be isolated during the late Pleistocene (Zhai et al. 2012). Therefore, we suggest that any intercontinental migration between Japan and Isl. Lanyu must have happened via the Ryukyu Islands bridge around early or middle Pleistocene when there was a strong connection between land masses (Kizaki and Oshiro 1977; Ota 1998; Fig. S1).
The paleo-vegetation reconstruction presented by Harrison et al. (2001) shows that the warm-temperate evergreen forests of East Asia were restricted to the southern region of East China and the southern tip of Japan along the Pacific coastal areas during the LGM. Those forests rapidly expanded and recovered northward from refugia during the post-glacial era. Several phylogeographical studies in this region have suggested southward range contractions during the LGM, followed by rapid northward expansions after de-glaciation (Aoki et al. 2004; Huang et al. 2002; Ohi et al. 2003a, b). However, our results demonstrate that isolated populations of N. sericea in East China (Isl. Putuo) and Taiwan (Isl. Lanyu) can be more plausibly explained by southward expansion from previously existing populations that served as northern refugia during the LGM or glacial periods.
Taxonomic entity of N. sericea var. aurata
Neolitsea sericea var. aurata was originally described by Hayata (1911) at Isl. Lanyu of Taiwan as a separate species, Litsea aurata. Subsequently, Hatusima (1969) considered the species to be a variety of N. sericea. That variety is distinguished from var. sericea in having dense, golden-brown hairs on the lower surfaces of its leaves. This treatment has been maintained (Chen and Pan 1997; Liao 1996; Miyazaki et al. 2003).
Our results revealed that N. sericea var. aurata and N. sericea var. sericea cannot be completely separated by molecular data because they share haplotype G. Nonetheless, our non-hierarchical AMOVA presents greater genetic divergence in contrasts that include Isl. Lanyu. In particular, the populations of Taiwan (Isl. Lanyu) and the Asiatic continent (Korean Peninsula and East China) show completely different lineages in the haplotype network. Furthermore, the population of var. aurata at Isl. Lanyu has a private haplotype H. Based on these findings, both the morphological and molecular evidence suggest that these two varieties have undergone a speciation event by geographical fragmentation. However, we cannot confirm the taxonomic position of var. aurata because we lack materials from the Ryukyu Islands, where that variety is also distributed (Hatusima 1969).
In conclusion, most of the haplotypes, including ancestral types, are found in the southwestern part of the Japanese main islands. Our genealogical analysis of haplotypes indicated that the putative basal group leads to two lineages in each of the other regions. The current distribution of N. sericea in East Asia has resulted from separate historical migrations through land bridges that connected the Japanese main islands to either the Korean Peninsula or Taiwan during the glacial periods. The Zhoushan populations have been shaped by post-glacial processes from northern refugia that existed during the late Pleistocene. Further work using additional genetic markers and including broad areas such as the Ryukyu Islands and southern China might provide a better understanding about the historical migration of evergreen tree species in East Asia.
References
Aoki K, Suzuki T, Hsu TW, Murakami N (2004) Phylogeography of the component species of broad-leaved evergreen forests in Japan, based on chloroplast DNA variation. J Plant Res 117:77–94
Aoki K, Matsumura T, Hattori T, Murakami N (2006) Chloroplast DNA phylogeography of Photinia glabra (Rosaceae) in Japan. Am J Bot 93:1852–1858
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Bai WN, Liao WJ, Zhang DY (2010) Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytol 188:892–901
Chen KC, Pan FJ (1997) New findings on the genus Neolitsea (Lauraceae) in Taiwan (in Chinese with English summary). Taiwan J For Sci 12:155–166
Chen KM, Abbott RJ, Milne RI, Tian XM, Liu JQ (2008) Phylogeography of Pinus tabulaeformis Carr. (Pinaceae), a dominant species of coniferous forest in northern China. Mol Ecol 17:4276–4288
Cheng YP, Hwang SY, Lin TP (2005) Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae). Mol Ecol 14:2075–2085
Chung MG, Chung MY, Oh GS, Epperson B (2000) Spatial genetic structure in a Neolitsea sericea population (Lauraceae). Heredity 85:490–497
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Corriveau JL, Coleman AW (1988) Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am J Bot 75:1443–1458
Duminil J, Fineschi S, Hampe A, Jordano P, Salvini D, Vendramin GG, Petit RJ (2007) Can population genetic structure be predicted from life-history traits? Am Nat 169:662–672
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Hampe A, Arroyo J, Jordano P, Petit RJ (2003) Rangewide phylogeography of a bird-dispersed Eurasian shrub: contrasting Mediterranean and temperate glacial refugia. Mol Ecol 12:3415–3426
Harrison SP, Yu G, Takahara H, Prentice IC (2001) Palaeovegetation: diversity of temperate plants in East Asia. Nature 413:129–130
Hatusima S (1969) On some species of the Lauraceae from Japan and Taiwan (in Japanese). J Geobot 17:106–107
Hayata B (1911) Materials for a flora of Formosa. Journal of the College of Science, Imperial University, Tokyo, Japan
Heuertz M, Fineschi S, Anzidei M, Pastorelli R, Salvini D, Paule L, Frascaria-Lacoste N, Hardy OJ, Vekemans X, Vendramin GG (2004) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13:3437–3452
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hewitt G (2003) Ice ages: species distributions, and evolution. In: Rothschild LJ, Lister AM (eds) Evolution on planet earth. Academic Press, Amsterdam
Hikida T, Ota H (1997) Biogeography of reptiles in the subtropical East Asian islands. In: Lue KY, Chen T-H (eds) Proceedings of the Symposium on the Phylogeny, Biogeography and Conservation of Fauna and Flora of East Asian Region. National Science Council, Taipei, pp 11–28
Hong SK, Nakagoshi N, Fu B, Morimoto Y (2007) Landscape ecological applications in man-influenced areas: linking man and nature systems. Springer, New York
Huang S, Chiang YC, Schaal BA, Chou CH, Chiang TY (2001) Organelle DNA phylogeography of Cycas taitungensis, a relict species in Taiwan. Mol Ecol 10:2669–2681
Huang SSF, Hwang SY, Lin TP (2002) Spatial pattern of chloroplast DNA variation of Cyclobalanopsis glauca in Taiwan and East Asia. Mol Ecol 11:2349–2358
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge
Kimura M (1996) Quaternary paleogeography of the Ryukyu Arc (in Japanese with English summary). J Geogr 105:259–285
Kimura M (2000) Paleography of the Ryukyu Islands. Tropics 10:5–24
Kitamura A, Kimoto K (2006) History of the inflow of the warm Tsushima Current into the Sea of Japan between 3.5 and 0.8 Ma. Palaeogeogr Palaeoclimatol Palaeoecol 236:355–366
Kizaki K, Oshiro I (1977) Paleogeography of the Rytkyu [sic] Islands (in Japanese with English summary). Marine Sci Mon 9:543–547
Kominami Y, Sato T, Takeshita K, Manabe T, Endo A, Noma N (2003) Classification of bird-dispersed plants by fruiting phenology, fruit size, and growth form in a primary lucidophyllous forest: an analysis, with implications for the conservation of fruit-bird interactions. Ornithol Sci 2:3–23
Lee JH, Choi BH (2010) Distribution and northernmost limit on the Korean Peninsula of three evergreen trees (in Korean with English summary). Korean J Pl Taxon 40:267–273
Lee JH, Lee BY, Choi BH (2011) Preliminary search of intraspecific chloroplast DNA variation of nine evergreen broad leaved plants in East Asia. Korean J Pl Taxon 41:194–201
Li EX, Yi S, Qiu YX, Guo JT, Comes HP, Fu CX (2008) Phylogeography of two East Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol Phylogenet Evol 49:702–714
Liao JC (1996) Neolitsea Merr. In: Editorial Committee of the Flora of Taiwan (eds) Flora of Taiwan, vol 2. Tah Jinn Printing Company Ltd., Taipei, pp 484–496
Mantel N (1967) The detection of disease clustering and generalized regression approach. Cancer Res 27:209–220
Miyazaki T, Yamaguchi YY, Hoshino Y, Ohba H (2003) Neolitsea sericea (Blume) Koidz. var. aurata (Hayata) Hatus. and the occurrence in Toshima, the Izu Islands, Japan (in Japanese with English summary). J Jpn Bot 78:311–312
Nei M (1987) Molecular evolutionary genetics. Oxford University Press, New York
Nishizawa T, Watano Y (2000) Primer pairs suitable for PCR-SSCP analysis of chloroplast DNA in angiosperms. J Phytogeogr Taxon 48:63–66
Ohashi H, Sasaki Y, Ohashi K (2006) The northern limit of distribution of Neolitsea sericea (Blume) Koidz. (Lauraceae) on the Pacific side of Japan (in Japanese with English summary). J Jpn Bot 81:248–249
Ohi T, Kajita T, Murata J (2003a) Distinct geographic structure as evidenced by chloroplast DNA haplotypes and ploidy level in Japanese Aucuba (Aucubaceae). Am J Bot 90:1645–1652
Ohi T, Wakabayashi M, Wu S, Murata J (2003b) Phylogeography of Stachyurus praecox (Stachyuraceae) in the Japanese Archipelago based on chloroplast DNA haplotypes. J Jpn Bot 78:1–14
Ota H (1998) Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their paleogeographical implications. Res Popul Ecol 40:189–204
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam BC, Deans JD, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Jensen JS, König AO, Lowe AJ, Madsen SF, Mátyás G, Munro RC, Popescu F, Slade D, Tabbener H, de Vries SMG, Ziegenhagen B, Beauliey JL, Kremer A (2002) Identification of refugia and post-glacial colonization routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manage 156:49–74
Petit RJ, Aguinagalde I, De Beaulieu J-L, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Pons O, Petit R (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Qian H, Ricklefs RE (2000) Large-scale processes and the Asian bias in species diversity of temperate plants. Nature 407:180–182
Qiu YX, Qi XS, Jin XF, Tao XY, Fu CX, Naiki A, Comes HP (2009a) Population genetic structure, phylogeography, and demographic history of Platycrater arguta (Hydrangeaceae) endemic to East China and South Japan, inferred from chloroplast DNA sequence variation. Taxon 58:1226–1241
Qiu YX, Sun Y, Zhang XP, Lee J, Fu CX, Comes HP (2009b) Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol 183:480–495
Qiu YX, Fu CX, Comes HP (2011) Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol Phylogenet Evol 59:225–244
Rendell S, Ennos R (2003) Chloroplast DNA diversity of the dioecious European tree Ilex aquifolium L. (English holly). Mol Ecol 12:2681–2688
Rozas J, Rozas R (1999) DNASP, version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15:174–175
Sugahara K, Kaneko Y, Ito S, Yamanaka K, Sakio H, Hoshizaki K, Suzuki W, Yamanaka N, Setoguchi H (2011) Phylogeography of Japanese horse chestnut (Aesculus turbinata) in the Japanese Archipelago based on chloroplast DNA haplotypes. J Plant Res 124:75–83
Taberlet P, Gielly L, Patou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wang ZS, An SQ, Liu H, Leng X, Zheng JW, Liu YH (2005) Genetic structure of the endangered plant Neolitsea sericea (Lauraceae) from the Zhoushan Archipelago using RAPD markers. Ann Bot 95:305–313
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci USA 84:9054–9058
Xie GW (1997) On phytogeographical affinities of the forest floras between East China and Japan. Chin Geogr Sci 19:789–799
Yiming L, Niemela J, Dianmo L (1998) Nested distribution of amphibians in the Zhoushan archipelago, China: can selective extinction cause nested subsets of species? Oecologia 113:557–564
Zhai SN, Comes HP, Nakamura K, Yan HF, Qiu YX (2012) Late Pleistocene lineage divergence among populations of Neolitsea sericea (Lauraceae) across a deep sea-barrier in Ryukyu Islands. J Biogeogr 37:1347–1360
Acknowledgments
The authors thank Emeritus Prof. H. Ohashi of Tohoku University, Dr. Y. Endo of Ibaraki University, Dr. T. Nemoto of Ishinomaki Senshu University, Dr. A. Yang of the National Museum of Natural Science, and Dr. S.H. Kang of Semyung University for help in collecting plant materials. They also thank all colleagues, J.Y. Lee, W.B. Cho, I.S. Choi, S.K. Jung, and D.P. Jin at the Plant Systematics Laboratory of Inha University for assisting with statistical analysis and commenting on the manuscript. This work was supported by Inha University and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2012R1A2A2A01011428).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10265_2012_519_MOESM1_ESM.tif
Fig. S1. Maps showing a hypothesis by Kizaki and Oshiro (1977), then modified according to that of Hikida and Ota (1997). a and b show inferred land configurations in East Asia during the early and middle Pleistocene, respectively (TIFF 4709 kb)
Rights and permissions
About this article
Cite this article
Lee, JH., Lee, DH. & Choi, BH. Phylogeography and genetic diversity of East Asian Neolitsea sericea (Lauraceae) based on variations in chloroplast DNA sequences. J Plant Res 126, 193–202 (2013). https://doi.org/10.1007/s10265-012-0519-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-012-0519-1