
Abstract
Several of the drugs currently used for the treatment of glomerular diseases are prescribed for their immunotherapeutic or anti-inflammatory properties, based on the current understanding that glomerular diseases are mediated by immune responses. In recent years our understanding of podocytic signalling pathways and the crucial role of genetic predispositions in the pathology of glomerular diseases has broadened. Delineation of those signalling pathways supports the hypothesis that several of the medications and immunosuppressive agents used to treat glomerular diseases directly target glomerular podocytes. Several central downstream signalling pathways merge into regulatory pathways of the podocytic actin cytoskeleton and its connection to the slit diaphragm. The slit diaphragm and the cytoskeleton of the foot process represent a functional unit. A breakdown of the cytoskeletal backbone of the foot processes leads to internalization of slit diaphragm molecules, and internalization of slit diaphragm components in turn negatively affects cytoskeletal signalling pathways. Podocytes display a remarkable ability to recover from complete effacement and to re-form interdigitating foot processes and intact slit diaphragms after pharmacological intervention. This ability indicates an active inside-out signalling machinery which stabilizes integrin complex formations and triggers the recycling of slit diaphragm molecules from intracellular compartments to the cell surface. In this review we summarize current evidence from patient studies and model organisms on the direct impact of immunosuppressive and supportive drugs on podocyte signalling pathways. We highlight new therapeutic targets that may open novel opportunities to enhance and stabilize inside-out pathways in podocytes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proper podocyte function and structure plays an important role in determining the integrity of the glomerular filtration barrier. Podocytes are highly differentiated epithelial cells with interdigitating foot processes that form a network on the outer surface of the filter slit diaphragms connecting neighbouring foot processes. Podocytes are terminally differentiated cells with no regenerative capacity. Therefore, a reduction in podocyte number beyond a critical threshold leads to glomerular disease progression in experimental rodent models [1, 2]. However, as long as the podocytes are just effaced and not lost, they display a remarkable capacity to recover foot processes within a short period of time, as occurs in minimal change disease, the classical example of the potential of podocytes to recover. However, effacement caused by other diseases also has the capability to recover, as described in a recent case of nephrotic range proteinuria following a Hantavirus infection which completely resolved within a few weeks [3].
Ultrastructural findings in this case report [3] document the cytoskeletal phenotype of foot process effacement as well as a redistribution of slit diaphragm proteins to the intracellular compartment of podocytes. This process clearly indicates that slit diaphragm components are not degraded immediately after internalization but may be transferred to intracellular storage compartments from where they can be recycled from the inside back out to the cell surface. Transient proteinuria models in rodents, such as the protamine sulphate model in rats and the lipopolysaccharide (LPS) model in mice display rapid but reversible foot process effacement and proteinuria. Even more striking, PS infusions in rodents can result in the fusion of podocyte foot processes, and this phenotype is reversible within minutes after infusion of heparin [4, 5]. Therefore, experimental evidence also suggests that a molecular machinery in podocytes exists to internalize slit diaphragm components and recycle them rapidly back to the cell surface. Most likely, as we recently demonstrated, those mechanisms are orchestrated by posttranslational modifications [6]. The targeting or supporting of these “inside-out” pathways could be a novel anti-proteinuric and podocyte-specific treatment concept. Other mechanisms of recovery involve glomerular cell replacement from stem cells. A glomerular stem cell niche was recently described which implies that lost podocytes could be replaced by cells from the parietal layer [7–9]. Whether this concept is only part of a physiological replenishment or holds true in disease states is a controversial discussion since parietal epithelial cells are clearly involved in the disease process itself [10–13].
In this review we summarize current evidence supporting the hypothesis that the established treatment regimens for nephrotic syndromes might be interfering directly with cellular pathways that stabilize and support the recovery of the actin cytoskeleton and the slit diaphragm of podocytes. If this hypothesis were to be valid, drugs commonly used in the clinic to treat our patients would have to be put into a new context, and there would be a need for novel and cell-specific therapeutic approaches since it most likely would be unnecessary to target the general immune system and accept the systemic side effects of immunosuppressive drugs.
The podocyte cytoskeleton plays a key role in proper glomerular function
A central aspect of studies in podocyte cell biology, which involves convergence of several signalling pathways of the deregulated immune system, is the actin cytoskeleton.
Podocyte foot processes consist of cortical actin filaments and actin-associated proteins, such as myosin, α-actinin and synaptopodin, which ensure the dynamic maintenance and reorganization of the cytoskeleton. The important role of the podocyte actin cytoskeleton and podocyte–glomerular basement membrane (GBM) interactions for the development of foot process effacement is supported by several genetic loss- or gain-of-function models affecting the cytoskeleton and recapitulating genetic human diseases [14–19]. It would therefore seem reasonable to search for supportive therapies that would directly interfere with pathways regulating slit diaphragm protein expression and cytoskeletal pathways that activate or support the recovery potential of stressed podocytes. In the following section we discuss widely subscribed drugs which have direct positive or negative (side-) effects on podocyte structures. Table 1 summarizes current knowledge.
Commonly prescribed drugs directly targeting the podocyte
Renin–angiotensin–aldosterone system blockers
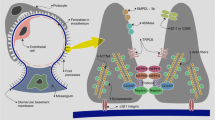
The renin–angiotensin–aldosterone (RAAS) system plays a crucial role in kidney disease. The major effector molecule of the RAAS is angiotensin II that mediates its function through angiotensin II type-1 and type-2 receptor (AT1-R and AT2-R). Immunofluorescence studies showed that both AT1-R and AT2-R are expressed in podocytes and that their expression is elevated in the proteinuric state [20]. Angiotensin II depolarizes podocytes directly through the opening of chloride ion channels and the resulting increased chloride conductance. The activation of chloride ion conductance is mediated by an AT1-R [21]. Angiotensin II infusion has been shown to induce proteinuria independent of any pressure effects [22] and to induce reorganization of the actin cytoskeleton and increase intracellular cAMP in cultured glomerular epithelial cells [23]. Durvasula et al. reported that exposure to cyclic stretch increased both the production of endogenous angiotensin II and the expression of AT1-R in cultured rat podocytes, and in vivo after 5/6 nephrectomy in rats [24]. In another study, the tissue RAAS of the kidneys was activated in diabetic nephropathy and immortalized murine podocytes, with higher concentrations of angiotensinogen, angiotensin II and AT1-R expressed under high glucose conditions [25].
RAAS blockers have beneficial effects on proteinuria and progression to renal failure. Lewis et al. showed a reno-protective effect of angiotensin-converting enzyme (ACE) inhibitors in type 1 diabetes [26]. RAAS blockers also have beneficial renal effects in type II diabetes [27]. The ACE inhibition in the Progressive Renal Insufficiency (AIPRI) study demonstrated that ACE inhibitors are able to delay the loss of renal function in non-diabetic patients.
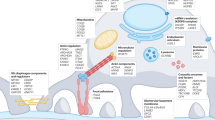
The anti-proteinuric effect of RAAS blockers is primarily thought to be mediated by a reduction in glomerular perfusion pressure. However, growing evidence suggests that RAAS inhibition might also act on the podocyte cytoskeleton and on components of the glomerular slit diaphragm directly. In podocytes RAAS inhibition results in decreased cell tonus, rearrangement of the actin cytoskeleton and decreased rate of apoptosis and protein leakage [24]. A direct effect of RAAS inhibition on the slit diaphragm is that it prevents redistribution of zona occludens 1 (ZO-1) in rats [28] and restores the expression of nephrin in experimental models of diabetic nephropathy [29]. Both of these proteins are important components for the proper filtration functioning of the glomerulus (Fig. 1). The selective aldosterone blocker eplerenone also ameliorates podocyte injury, proteinuria and salt-evoked nephropathy [30, 31].

Illustration of different drug targets in the podocyte. Positive effects of drugs on podocyte function are shown in green and negative effects of drugs on podocyte function are shown in red. Please note that mammalian target of rapamycin (mTOR) inhibitors are mentioned in both colours as they have pro-survival effects in diabetic nephropathy but can cause negative side effects on podocytes in other clinical settings. Here mTOR inhibitors can lead to proteinuria and podocyte apoptosis by decreasing the expression of synaptopodin, podocin and nephrin. ARBs Angiotensin receptor blocker, ASMase acid sphingomyelinase, AT1-R angiotensin 1 receptor, CD2AP CD2-associated protein, CTLA-4 cytotoxic T lymphocyte-associated protein 4, MC-R mineralocorticoid receptor, PPARγ peroxisome proliferator-activated receptors γ, SMPDL 3b sphingomyelin phosphodiesterase acid-like 3b protein, SUMOs small ubiquitin-like modifiers, SYNPO synaptopodin, suPAR soluble urokinase plasminogen activator receptor, TGF-R transforming growth factor receptor, TRPC-6 transient receptor potential cation channel 6, VEGF-R vascular endothelial growth factor receptor
Glucocorticoids
The current paradigm is that glucocorticoids act in glomerular disease by reducing the release of pathogenic factors of circulating T lymphocytes that are thought to be one of the major causes of some forms of idiopathic nephrotic syndromes. Glucocorticoids bind to the cytoplasmic glucocorticoid receptor that is translocated to the nucleus after dimerization. In the nucleus glucocorticoids bind to glucocorticoid response elements on the DNA or interact with other transcription factors [32]. However, glucocorticoids might have important direct effects on the cells as well (Fig. 1). Human podocytes also express the glucocorticoid receptors [33] and in vitro, dexamethasone treatment protects podocytes from puromycin aminonucleoside (PAN) injury by inhibiting actin filament disruption and PAN-induced apoptosis [34, 35]. Similar to the PAN model, dexamethasone can also rescue podocytes from adriamycin-induced actin rearrangement by stabilizing the expression of α-actinin-4 [36].
In a differential proteomic analysis of dexamethasone-treated cultured murine podocytes ciliary neurotrophic factor, αB-crystallin and heat shock protein 27 were upregulated by dexamethasone. These three proteins play a well-known role in protecting cells from injury, and current data suggest a direct effect of steroids on podocytes in nephrotic diseases [34]. Most of the in vivo studies of experimental nephrotic syndrome induced in rats by PAN-injection performed to date have demonstrated that glucocorticoid treatment reduces proteinuria and attenuates podocyte foot process effacement [37, 38]. In humans, glucocorticoid therapy remains the primary treatment option for nephrotic syndrome, and in many cases of minimal change disease, steroid treatment induces remission and restoration of the slit diaphragm architecture, leading to the term “steroid-sensitive nephrotic syndrome” [39]. Steroid-resistant cases usually receive combination treatments with addition of calcineurin inhibitors (CNIs).
Calcineurin inhibitors
Calcineurin inhibitors (CNIs), such as cyclosporine A or Fk506, lower the activity of T cells. The immunosuppressive actions of cyclosporine were originally attributed to the de-phosphorylation of the nuclear factor of activated T cell (NFAT) family members (Fig. 2) as CNIs lead to nuclear translocation and activation of early genes of the T cell-driven immune response [40]. However, Faul et al. showed that the beneficial effect of cyclosporine A on proteinuria is not dependent on NFAT inhibition in T cells. In podocytes, calcineurin dephosphorylates synaptopodin and makes it more susceptible to cathepsin L-mediated degradation. By inhibiting calcineurin, cyclosporine A stabilizes the actin cytoskeleton in podocytes by preserving the phosphorylation-dependent synaptopodin-14-3-3β integrin interaction [41] (Fig. 1). In addition to the effectiveness in glomerular diseases that are thought to be immunologically mediated, CNIs are also used to reduce proteinuria in Alport syndrome, which is a clear non-immunological disease caused by mutations of the type IV collagen of the GBM [42–44], further supporting the notion of additional CNI-mediated effects independent of T cells. Additional evidence derives from studies in children with genetic podocytopathies. Here cyclosporine A was demonstrated to have an anti-proteinuric effect in cases with mutations in the WT-1, podocin and phospholipase C epsilon genes [45–47].

Signalling pathways activated by different drugs in podocytes. AKT Protein kinase B, Ang I/II angiotensin I/II, DG diacylglycerol, ERK extracellular signal-regulated kinases, IP3 inositol trisphosphate 3, JAK janus kinase, MAK mitogen-activated protein kinases, NFAT nuclear factor of activated T-cells, PI3K phosphatidylinositol-4,5-bisphosphate 3-kinase, PKC-α protein kinase C alpha, PLC phospholipase C, STAT signal transducer and activator of transcription, TSC tuberous sclerosis complex. For other abbreviations, see caption to Fig. 1
Rituximab
Rituximab is a monoclonal antibody directed against the CD20 receptor expressed on B lymphocytes but has also been demonstrated to be effective in glomerular diseases that are not related to B cells, such as recurrent focal segmental glomerulosclerosis (FSGS) after transplantation [48]. As recently reported by Ruggenenti et al., rituximab can also reduce the number of relapses in patients with minimal change disease (MCD) and FSGS [49]. Interestingly, rituximab binds not only to CD20 but also to other molecules, including sphingomyelin phosphodiesterase acid-like 3b protein (SMPDL-3b), in immune cells [50]. Interestingly, the authors of a recent study suggested that rituximab controls actin cytoskeleton remodelling in podocytes through the stabilization of sphingolipid-related enzymes [51] (Fig. 1). In this study, SMPDL-3b downregulation after exposure to FSGS patient sera rendered podocytes more susceptible to actin remodelling, and rituximab partially preserved the disruption of stress fibers through the stabilization of SMPDL-3b [51].
Mammalian target of rapamycin inhibitors
Mammalian target of rapamycin (mTOR) is a serine–threonine kinase, which controls cell growth and metabolism. MTOR also has a central role in cell survival. It mediates its functions through mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) as well as through regulatory-associated protein of mTOR (Raptor) and rapamycin-insensitive companion of mTOR (Rictor) [52]. Moreover, mTOR regulates vascular endothelial growth factor (VEGF) that is essential for podocyte survival through autocrine and paracrine pathways [53]. Current data on the function of mTOR inhibitors in renal disease are conflicting.
It has been shown that hyperactivation of the mTOR pathway in diabetic nephropathy plays a pivotal role in the hypertrophy of glomerular cells and is associated with podocyte injury and the progressive decline of glomerular filtration rates [54] and that inhibition of the mTORC1 pathway with rapamycin has reno-protective effects on the progression of diabetic nephropathy [55]. However, the induction of proteinuria after treatment with mTOR inhibitors, such as sirolimus and everolimus, is a typical side effect [56, 57]. In a knock-out model Raptor deletion induced proteinuria and glomerular lesions in mice, and concomitant Rictor deletion exacerbated this phenotype [52, 58, 59]. Sirolimus has also been shown to reduce the expression of synaptopodin, podocin and nephrin and in addition to increase podocyte apoptosis [60]. VEGF expression was reduced in the glomeruli of patients with sirolimus-induced thrombotic microangiopathy [61]. Thus, mTOR inhibitors seem to have direct effects on podocyte integrity. However, on the one hand mTOR inhibitors have the side effect of proteinuria; on the other hand they have been shown to have beneficial effects in diabetic nephropathy. The conflicting data of mTOR regarding glomerular function might indicate that a delicate balance of mTOR and/or subtype-specific receptor activity is necessary for proper podocyte function. In this regard mTOR function seems to be similar to VEGF function (see below) that also requires an orchestrated activity in the glomerulus. To date it is not clear whether there is a specific mTOR pathway that should be targeted depending on the disease that should be treated. More studies on mTOR subtype-specific functions are needed.
3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors—also known as statins—are a class of drugs used to lower cholesterol levels by inhibiting the enzyme HMG-CoA reductase that plays a central role the production of cholesterol in the liver. HMG-CoA reductase inhibitors have been shown to decrease the rate of decline of kidney function and to reduce proteinuria in patients with chronic kidney disease stages I–III [62, 63]. These effects are independent of serum lipid levels, implicating pleiotropic effects of statins in the kidney [64]. In addition to their general effect on lipid metabolism, statins seem to have beneficial effects on mitochondrial function in podocytes. Rosuvastatin attenuated angiotensin II-dependent increases in NADPH oxidase activity and reactive oxygen species (ROS) generation in cultured podocytes [65]. In line with these findings are reports that fluvastatin is able to attenuate injury induced by PAN and to increase the production of β1-integrin in human podocytes in vitro, which is thought to be an ROS activity-inhibiting mechanism [66, 67]. Rosuvastatin has also been found to display pro-survival activities in injured podocytes through a p21-dependent anti-apoptotic pathway [68].
Peroxisome proliferator-activated receptors γ agonists
Thiazolidinediones (TZDs)—also known as glitazones—are anti-diabetic agents which activate the peroxisome proliferator-activated receptors γ (PPARγ), leading to modified gene transcription that improves insulin resistance, among others. Growing evidence suggests that TZDs also may affect PPARγ-independent pathways in glomerular disease. TZDs have been shown to reduce podocyte injury and proteinuria in diabetic nephropathy [69, 70]. In two studies, the development of PAN-induced glomerulosclerosis could be ameliorated by pioglitazone which also reduced apoptosis and necrosis in cultured podocytes [70, 71]. Similar, rosiglitazone was observed to attenuate the development of proteinuria and glomerulosclerosis in doxorubicin-induced FSGS in rats [72]. Rosiglitazone is able to protect podocytes against damage caused by mitochondrial dysfunction [73]. Moreover, TZDs can prevent actin filament redistribution induced by PAN [74] and directly influence expression of small ubiquitin-like modifiers (SUMOs) [75], which we recently demonstrated are essential for the cellular surface expression of nephrin in podocytes (Fig. 1) [6].
Thus, PPARγ agonists do not only lower blood glucose levels in diabetes but are also beneficial in diabetic nephropathy by improving podocyte mitochondrial function, slit diaphragm integrity and stability of the actin cytoskeleton.
VEGF inhibitors
Podocytes are the major source of VEGF production in the glomerulus. We and others have shown that podocytes express different VEGF isoforms and also VEGF receptors (VEGF-R) [76, 77]. Furthermore, VEGF-A and VEGF-C have an important autocrine role in the podocyte, and VEGF inhibition leads to the activation of pro-apoptotic pathways in cultured human podocytes [77]. Podocyte-specific heterozygous and homozygous deletions of VEGF results in proteinuria and endotheliosis in mice [78]. VEGF also has direct beneficial effects on podocyte survival independent of nephrin expression [79]. An endogenous condition when VEGF-A levels are reduced is preeclampsia. Here a soluble VEGF-R1 (sFLT-1) produced by the placenta blocks VEGF in the circulation and causes proteinuria, hypertension and endotheliosis [80, 81]. Serum from women with preeclampsia and with high sFlt-1 concentrations can directly alter podocyte structure and function whereas supplementing these sera with exogenous VEGF can directly reverse is effects [82]. These studies clearly indicate that podocyte function depends on a certain level of autocrine VEGF-R activation. The delivery of VEGF121 via the tail vein normalizes VEGF levels and prevents the late-gestational spike in blood pressure and proteinuria in a murine model for preeclampsia [83]. In a rat model for preeclampsia the treatment with VEGF121 reverses hypertension, proteinuria and glomerular endotheliosis [84]. To our knowledge, VEGF-A treatment has not yet been used in humans to treat proteinuria. However, because of the negative effects seen in diabetic nephropathy (see below) more work has to be done to elucidate more clearly the precise tuning of VEGF amount and different isoforms before VEGF infusion can be tried in patients. Based on our own work treatment with VEGF-C could be a short-term alternative [77].
The levels of VEGF, while downregulated in preeclampsia VEGF levels, are upregulated in diabetic nephropathy. Moreover, podocyte VEGF164 overexpression in mice is sufficient to induce structural and functional abnormalities in the glomerular filtration barrier similar to those in diabetic nephropathy [85]. Anti-VEGF antibodies reduce the severity of diabetic nephropathy in rodents [86, 87], whereas they can induce hypertension, proteinuria and glomerular disease in human patients [88]. Anti-VEGF strategies that either block the extracellular binding of VEGF to its receptor (anti-VEGF antibodies) or inhibit intracellular signalling pathways of VEGF-R (receptor tyrosine kinase inhibitors) are widely used in the clinical setting to inhibit angiogenesis in different types of cancer. In patients treatment with VEGF inhibitors can lead to podocyte effacement. Our group was the first to describe podocyturia (loss of podocytes in the urine) in patients receiving VEGF ablation therapy with bevacizumab or sunitinib [89].
Proteinuria recovery following the discontinuation of VEGF ablation therapy indicates the direct beneficial effect of this treatment and underlines the remarkable recovery potential of podocytes.
The conflicting data on the VEGF system in different species and different glomerular diseases suggest that a dynamic and context-related balance of VEGF levels and isoforms is necessary to maintain proper glomerular function. Consequently, this molecule is a difficult potential drug target.
New strategies to directly target the podocyte
Notch inactivation
Therapies based on newly identified podocyte proteins and signalling pathways might be the future for the treatment of glomerular disease. For example, inactivation of Notch signalling pathways in podocytes, which promotes apoptosis, could be a promising target pathway to ameliorate the damage caused by glomerular disease [90]. A major problem with this therapeutic strategy is that many of the pathways which are upregulated or reactivated in glomerular disease might be essential for maintaining homeostasis in other tissues. Therefore, a more podocyte-directed therapy is highly desirable. In the following sections we present new therapeutic strategies by directly targeting the podocyte. Table 2 summarizes the effects of new anti-proteinuric strategies that potentially target podocyte structures and pathways.
Abatacept
Abatacept (CTLA-4–Ig) consists of a fusion protein of the extracellular domain of CTLA-4 and human immunoglobulin (Ig) G1. CTLA-4 transmits an inhibitory signal to T cells and through its binding to CD80 (also called B7-1) on antigen-presenting cells, it prevents the delivery of the co-stimulatory signal to the T cell [91, 92]. Abatacept is used in glomerular disease to suppress the T cell activity that is thought to be a major cause in some of these diseases. For example, MCD is commonly thought to be mediated by a circulating factor released by T cells. However, a couple of years ago an unanticipated novel role for CD80 in podocytes as an inducible modifier of glomerular permselectivity was described [4]. While CD80 expression is absent in normal podocytes, CD80 was found to be induced in podocytes in various animal models of proteinuria and was detected in podocytes from patients with glomerular diseases.
Increased podocytic CD80 expression has been found in genetic, drug-induced, immune-mediated and bacterial toxin–induced experimental nephrotic syndrome [4]. The exposure of podocytes to LPS rapidly unregulate CD80 in mice and leads to actin reorganization and nephrotic-range proteinuria, whereas mice lacking CD80 are protected from this damage [4]. Sera from MCD patients in relapse, but not in remission, were found to stimulate CD80 expression in cultured podocytes [93]. In addition, urinary concentrations of soluble CD80 in patients with relapsed MCD were significantly higher than those in patients with MCD in remission and with other glomerular diseases including FSGS and in healthy controls [94]. Serum and urinary CTLA-4 levels tend to be low in MCD patients in relapse, and the urinary CTLA-4 level returns to higher levels during remission [95]. Yu et al. described the use of abatacept in inducing remission in five patients with FSGS based on positive staining for CD80 in glomerular podocytes, reporting a clinically significant reduction in proteinuria in all patients [96]. However, the validity of these findings were questioned by other researchers [97, 98]. An ongoing clinical phase III trial that is evaluating abatacept for the treatment of lupus class III and IV is also analysing the outcome of proteinuria (https://clinicaltrials.gov/ct2/show/NCT01714817).
Soluble form of the urokinase plasminogen activator receptor blockers
Another potential therapeutic option might be the soluble form of the urokinase plasminogen activator receptor (suPAR) blocker. SuPAR secretion by T lymphocytes has been suggested to be a “permeability factor” in the pathogenesis of FSGS. It binds to α5β3 integrin in the podocyte membrane, thereby leading to podocyte contraction and loss of podocytes from the GBM [99, 100]. Amiloride inhibits the synthesis of uPAR and suPAR secretion by T lymphocytes and thereby decreases α5β3 integrin activation [101, 102].
However, the role of suPAR in glomerular disease has been questioned in recent years. Even though experimental data suggest that suPAR therapy may also cause proteinuria and FSGS by alternated β3-integrin signalling, these findings cannot be easily translated to routine clinical care [103]. Higher serum or plasma suPAR levels have also been demonstrated in patients with other diseases, such as cancer, sepsis and atherosclerosis [104, 105]. Future studies are ongoing to specifically reduce suPAR levels in patients with primary or recurrent FSGS.
Transient receptor potential cation channel 6 small interfering RNA
Another exciting concept is the direct and specific targeting of therapeutic agents to the podocyte, thereby limiting systemic adverse effects. One such example has been described for the transient receptor potential cation channel 6 (TRPC6). TRPCs are non-selective cationic channels which play a major role in chemo- and mechanosensation. In podocytes, TRPC6 is a slit diaphragm-interacting protein [106] which may regulate changes in calcium ion (Ca2+) levels and in actin cytoskeleton rearrangement [107]. Overexpression of TRPC6 in mice induces glomerular disease [108]. In humans gain-of-function mutations in TRPC6 have been associated with familial forms of FSGS [109]. Novel therapeutic options might involve small interfering RNAs (siRNAs) coupled with a podocyte-specific delivery system. A TRPC6 siRNA coupled with a sheep anti-mouse podocyte antibody has already been shown to reduce TRPC6 protein expression in podocytes: the IgG–siRNA complex was endocytosed into the podocyte and interfered with protein expression [110]. To date, there has been no clinical trial with TRPC6 siRNA.
Anti-transforming growth factor beta therapy
Transforming growth factor beta-1 (TGF-β1) and Smad7 induce apoptosis in podocytes through different downstream pathways leading to podocyte depletion and progressive glomerulosclerosis [111]. In recent years case reports have shown a reduction of proteinuria in patients receiving anti-TGF-β therapy. TNF-α blockade was successfully used to treat nephrotic syndrome in a patient with AA amyloid due to TNF receptor-associated periodic syndrome [112], and complete reversal of nephrotic syndrome secondary to amyloidosis was observed in a patient with inflammatory bowel disease and ankylosing spondylitis after treatment with infliximab (anti-TNF-α antibody) [113]. In a small study with 15 patients with renal amyloidosis, anti-TNF therapy had positive effects on the reduction of proteinuria in some patients [114]. Alternatively, Stokes et al. reported five patients with long-term rheumatoid arthritis who developed new-onset glomerular disease while receiving TNF-α antagonist. The authors suggested an induction of rheumatoid arthritis-related nephropathy or de novo autoimmune disorders due to the anti-TNF therapy [115].
The aim of a phase II trial carried out between 2008 and 2014 was to test whether adalimumab and/or galactose can safely reduce proteinuria and protect kidney function better than standard treatment for patients with FSGS (https://clinicaltrials.gov/ct2/show/ NCT00814255). The study has been completed, but the results are not yet available.
Oral N-acetyl-D-mannosamine
Sialic acids are essential for a variety of cellular functions, including cell adhesion and signal recognition as well as the formation and progression of tumors. Disruption of sialic acids can result in severe proteinuria. N-acetyl-D-mannosamine (ManNAc) is the precursor of all physiological sialic acids. The discovery of a central, mechanistic role played by two different forms of angiopoietin-like 4 (Angptl 4) in human and experimental glomerular disease has opened new treatment avenues. Localized upregulation of a hyposialylated form of glycoprotein Angptl 4 secreted by podocytes induces the cardinal features of human MCD, suggesting that glycoprotein Angptl 4 upregulation is a significant contributor toward proteinuria in experimental diabetic nephropathy. Oral treatment with ManNAc improves sialylation of Angptl 4 in vivo and reduces proteinuria by over 40 % [116]. Oral ManNAc is currently being tested in two phase I clinical trial for the treatment of the rare disorder distal myopathy with rimmed vacuoles–hereditary inclusion body myopathy (http://clinicaltrials.gov/NCT01236898 and NCT01359319) [117]. To our knowledge, there is no ongoing study involving ManNAc in patients with glomerular disease.
Isoform-specific protein kinase C inhibitors
Among various podocyte kinases, proper protein kinase C (PKC) signalling plays a critical role in podocyte function. Where some PKC isoforms are indispensable for proper glomerular development, others might be harmful to the glomerulus when activated in diabetes [118]. Moreover, a combined deletion of atypical PKC-λ/ι and atypical PKC-ζ isoforms in podocytes is associated with incorrectly positioned centrosomes and Golgi apparatus and mis-localized molecules of the slit diaphragm, leading to proteinuria [14]. Menne et al. reported that diabetic PKC-α isoform-null mice are resistant to albuminuria and glomerular upregulation of the VEGF system [119].
The lack of PKC-β can provide protection against diabetes-induced renal hypertrophy and glomerular hyperfiltration. [120]. The ability of isoform-specific PKC inhibitors to antagonize diabetes-induced glomerulonephropathy might be a new avenue for future therapeutic options. A selective PKC-β inhibitor (ruboxistaurin) has been used in clinical trials to determine its efficacy in diabetic macular edema, retinopathy, neuropathy, nephropathy and endothelial dysfunction in patients with type I or type II diabetes. The results of a phase II clinical trial suggest that ruboxistaurin can decrease the loss of glomerular filtration rate and proteinuria in diabetic patients who are already receiving as therapy the optimal RAAS blockade [121].
Protease inhibitors
It has been suggested that alterations in the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)/IκBα (inhibitor of kappa B) regulatory feedback loop contribute to immunologic abnormalities observed in MCD [122]. NF-κB is centrally regulated by the proteasome. Saquinavir is a protease inhibitor usually used to treat human immunodeficiency virus. In a small case series of patients with steroid-dependent and steroid-resistant nephrotic syndrome, the addition of saquinavir to therapy with other immunosuppressive drugs reduced proteinuria and had a steroid-sparing effect [123]. These results give hope to a new management strategy for nephrotic patients.
Conclusions
For years nephrologists have successfully used immunosuppressive therapy for the treatment of glomerular diseases, based on the rationale that many glomerular diseases are autoimmune-mediated. However, recent research has revealed that many of these drugs have direct effects on the podocyte - mostly through alterations of the actin cytoskeleton. Figures 1 and 2 summarize our current understanding of how drugs directly act on podocytes and inform the affected downstream signalling pathways. This information is of special importance because in some cases podocyte injury can be reversible, and the actin cytoskeleton has the ability to reorganize and restore interdigitating foot processes and recycle slit diaphragm proteins back to the cell surface.
The roles of the mTOR and the VEGF signalling pathways in this context are controversial. It now appears that both pathways require a fine-tuned, balanced baseline activation to maintain glomerular physiology. Thus, the question of whether parts of these signalling cascades can be safely targeted, and if so which parts, remains to be answered.
A close crosstalk between researchers and clinicians is of particular importance for the development of novel podocyte-specific drugs and for the identification of podocyte-specific drug targets in addition to those already widely used to treat glomerular disease, with the aim to minimize the general side effects of therapy in our patients.
Key points
-
1.
A crucial structure for podocyte maintenance is the actin cytoskeleton.
-
2.
It is apparent that many of the drugs currently used to treat glomerular diseases have direct effects on the podocytes and their cytoskeleton, but are still not podocyte-specific.
-
3.
Some drugs have direct side effects on podocytes, causing proteinuria and loss of podocytes from the GBM.
-
4.
The discovery of novel genes and signalling pathways involved in glomerular diseases will facilitate the future development of podocyte-specific drugs.
Multiple choice questions (answers are provided following the reference list)
-
1.
Which of the following answers is correct
-
a)
The mTOR pathway is inactivated in diabetes.
-
b)
mTOR inhibitors can induce proteinuria.
-
c)
mTOR inhibitors have only immune modulatory functions.
-
d)
mTOR inhibitors bind to special receptors on the podocyte surface.
-
a)
-
2.
Which of the following proteins is neither a unit of the podocyte cytoskeleton nor has a direct interaction with it?
-
a)
Actin
-
b)
Synaptopodin
-
c)
ZO-1
-
d)
Nephrin
-
a)
-
3.
Which of the following is a podocyte-specific drug?
-
a)
Cyclosporine
-
b)
Prednisolone
-
c)
Rituximab
-
d)
mTOR inhibitors
-
e)
None of the above
-
a)
-
4.
Which of the following statements is correct
-
a)
Human podocytes express CD20.
-
b)
Human podocytes do not express the angiotensin II type 1 receptor
-
c)
Human podocytes express the glucocorticoid receptor
-
d)
Human podocytes express the abatacept-binding partner under physiological conditions.
-
a)
-
5.
Which statement is correct?
-
a)
VEGF inhibitors only have renal side effects on the glomerular endothelium
-
b)
Bevacizumab has a protective effect on podocytes
-
c)
VEGF inhibitors can lead to foot process effacement
-
d)
VEGF receptors are only expressed on glomerular endothelial cells
-
a)
References
Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R (2001) Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60:957–968
Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC (2005) Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16:2941–2952
Boehlke C, Hartleben B, Huber TB, Hopfer H, Walz G, Neumann-Haefelin E (2014) Hantavirus infection with severe proteinuria and podocyte foot-process effacement. Am J Kidney Dis 64:452–456
Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, Faul C, Kretzler M, Davidson A, Sugimoto H, Kalluri R, Sharpe AH, Kreidberg JA, Mundel P (2004) Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest 113:1390–1397
Seiler MW, Venkatachalam MA, Cotran RS (1975) Glomerular epithelium: structural alterations induced by polycations. Science 189:390–393
Tossidou I, Himmelseher E, Teng B, Haller H, Schiffer M (2014) SUMOylation determines turnover and localization of nephrin at the plasma membrane. Kidney Int 86:1161–1173
Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ (2009) Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20:333–343
Shankland SJ, Anders HJ, Romagnani P (2013) Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr Opin Nephrol Hypertens 22:302–309
Lasagni L, Romagnani P (2010) Glomerular epithelial stem cells: the good, the bad, and the ugly. J Am Soc Nephrol 21:1612–1619
Berger K, Moeller MJ (2014) Podocytopenia, parietal epithelial cells and glomerulosclerosis. Nephrol Dial Transplant 29:948–950
Smeets B, Moeller MJ (2012) Parietal epithelial cells and podocytes in glomerular diseases. Semin Nephrol 32:357–367
Moeller MJ, Smeets B (2014) Role of parietal epithelial cells in kidney injury: the case of rapidly progressing glomerulonephritis and focal and segmental glomerulosclerosis. Nephron Exp Nephrol 126:97
Achenbach J, Mengel M, Tossidou I, Peters I, Park JK, Haubitz M, Ehrich JH, Haller H, Schiffer M (2008) Parietal epithelia cells in the urine as a marker of disease activity in glomerular diseases. Nephrol Dial Transplant 23:3138–3145
Hartleben B, Widmeier E, Suhm M, Worthmann K, Schell C, Helmstadter M, Wiech T, Walz G, Leitges M, Schiffer M, Huber TB (2013) aPKClambda/iota and aPKCzeta contribute to podocyte differentiation and glomerular maturation. J Am Soc Nephrol 24:253–267
Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, Walz G, Haller H, Schiffer M (2009) Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol 20:798–806
Worthmann K, Leitges M, Teng B, Sestu M, Tossidou I, Samson T, Haller H, Huber TB, Schiffer M (2013) Def-6, a novel regulator of small GTPases in podocytes, acts downstream of atypical protein kinase C (aPKC) lambda/iota. Am J Pathol 183:1945–1959
Henderson JM, Alexander MP, Pollak MR (2009) Patients with ACTN4 mutations demonstrate distinctive features of glomerular injury. J Am Soc Nephrol 20:961–968
Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, Wu G, Homstad A, Sparks MA, Gomez J, Jiang R, Alonso A, Lavin P, Conlon P, Korstanje R, Stander MC, Shamsan G, Barua M, Spurney R, Singhal PC, Kopp JB, Haller H, Howell D, Pollak MR, Shaw AS, Schiffer M, Winn MP (2014) Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol 25:1991–2002
Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS (1999) Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286:312–315
Suzuki K, Han GD, Miyauchi N, Hashimoto T, Nakatsue T, Fujioka Y, Koike H, Shimizu F, Kawachi H (2007) Angiotensin II type 1 and type 2 receptors play opposite roles in regulating the barrier function of kidney glomerular capillary wall. Am J Pathol 170:1841–1853
Gloy J, Henger A, Fischer KG, Nitschke R, Mundel P, Bleich M, Schollmeyer P, Greger R, Pavenstadt H (1997) Angiotensin II depolarizes podocytes in the intact glomerulus of the rat. J Clin Invest 99:2772–2781
Lapinski R, Perico N, Remuzzi A, Sangalli F, Benigni A, Remuzzi G (1996) Angiotensin II modulates glomerular capillary permselectivity in rat isolated perfused kidney. J Am Soc Nephrol 7:653–660
Sharma R, Lovell HB, Wiegmann TB, Savin VJ (1992) Vasoactive substances induce cytoskeletal changes in cultured rat glomerular epithelial cells. J Am Soc Nephrol 3:1131–1138
Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ (2004) Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 65:30–39
Yoo TH, Li JJ, Kim JJ, Jung DS, Kwak SJ, Ryu DR, Choi HY, Kim JS, Kim HJ, Han SH, Lee JE, Han DS, Kang SW (2007) Activation of the renin-angiotensin system within podocytes in diabetes. Kidney Int 71:1019–1027
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (1993) The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. the collaborative study group. N Engl J Med 329:1456–1462
Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, RENAAL Study Investigators (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345:861–869
Macconi D, Ghilardi M, Bonassi ME, Mohamed EI, Abbate M, Colombi F, Remuzzi G, Remuzzi A (2000) Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J Am Soc Nephrol 11:477–489
Davis BJ, Cao Z, de Gasparo M, Kawachi H, Cooper ME, Allen TJ (2003) Disparate effects of angiotensin II antagonists and calcium channel blockers on albuminuria in experimental diabetes and hypertension: potential role of nephrin. J Hypertens 21:209–216
Nagase M, Fujita T (2008) Aldosterone and glomerular podocyte injury. Clin Exp Nephrol 12:233–242
Navaneethan SD, Nigwekar SU, Sehgal AR, Strippoli GF (2009) Aldosterone antagonists for preventing the progression of chronic kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol 4:542–551
Muller M, Renkawitz R (1991) The glucocorticoid receptor. Biochim Biophys Acta 1088:171–182
Yan K, Kudo A, Hirano H, Watanabe T, Tasaka T, Kataoka S, Nakajima N, Nishibori Y, Shibata T, Kohsaka T, Higashihara E, Tanaka H, Watanabe H, Nagasawa T, Awa S (1999) Subcellular localization of glucocorticoid receptor protein in the human kidney glomerulus. Kidney Int 56:65–73
Ransom RF, Vega-Warner V, Smoyer WE, Klein J (2005) Differential proteomic analysis of proteins induced by glucocorticoids in cultured murine podocytes. Kidney Int 67:1275–1285
Yu-Shengyou LY (2013) Dexamethasone inhibits podocyte apoptosis by stabilizing the PI3K/Akt signal pathway. Biomed Res Int 2013:326986
Liu H, Gao X, Xu H, Feng C, Kuang X, Li Z, Zha X (2012) Alpha-actinin-4 is involved in the process by which dexamethasone protects actin cytoskeleton stabilization from adriamycin-induced podocyte injury. Nephrology (Carlton) 17:669–675
Fujiwara Y (1984) An ultrastructural study of the effect of the steroid in puromycin aminonucleoside nephrosis rats. Virchows Arch A Pathol Anat Histopathol 405:11–24
Trachtman H, Del Pizzo R, Valderrama E, Gauthier B (1990) The renal functional and structural consequences of corticosteroid and angiotensin-converting enzyme inhibitor therapy in chronic puromycin aminonucleoside nephropathy. Pediatr Nephrol 4:501–504
Waldman M, Crew RJ, Valeri A, Busch J, Stokes B, Markowitz G, D’Agati V, Appel G (2007) Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol 2:445–453
Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109[Suppl]:S67–79
Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P (2008) The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14:931–938
Charbit M, Gubler MC, Dechaux M, Gagnadoux MF, Grunfeld JP, Niaudet P (2007) Cyclosporin therapy in patients with alport syndrome. Pediatr Nephrol 22:57–63
Chen D, Jefferson B, Harvey SJ, Zheng K, Gartley CJ, Jacobs RM, Thorner PS (2003) Cyclosporine a slows the progressive renal disease of alport syndrome (X-linked hereditary nephritis): results from a canine model. J Am Soc Nephrol 14:690–698
Callis L, Vila A, Carrera M, Nieto J (1999) Long-term effects of cyclosporine A in alport’s syndrome. Kidney Int 55:1051–1056
Bensman A, Niaudet P (2010) Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr Nephrol 25:1197–1199
Gellermann J, Stefanidis CJ, Mitsioni A, Querfeld U (2010) Successful treatment of steroid-resistant nephrotic syndrome associated with WT1 mutations. Pediatr Nephrol 25:1285–1289
Malina M, Cinek O, Janda J, Seeman T (2009) Partial remission with cyclosporine A in a patient with nephrotic syndrome due to NPHS2 mutation. Pediatr Nephrol 24:2051–2053
Ponticelli C (2010) Recurrence of focal segmental glomerular sclerosis (FSGS) after renal transplantation. Nephrol Dial Transplant 25:25–31
Ruggenenti P, Ruggiero B, Cravedi P, Vivarelli M, Massella L, Marasa M, Chianca A, Rubis N, Ene-Iordache B, Rudnicki M, Pollastro RM, Capasso G, Pisani A, Pennesi M, Emma F, Remuzzi G, Rituximab in Nephrotic Syndrome of Steroid-Dependent or Frequently Relapsing Minimal Change Disease Or Focal Segmental Glomerulosclerosis (NEMO) Study Group (2014) Rituximab in steroid-dependent or frequently relapsing idiopathic nephrotic syndrome. J Am Soc Nephrol 25:850–863
Perosa F, Favoino E, Caragnano MA, Dammacco F (2006) Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross-reactivity with acid sphingomyelinase-like phosphodiesterase 3b precursor. Blood 107:1070–1077
Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, 3rd Burke GW (2011) Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 3:85ra46
Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mor A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstadt H, Kerjaschki D, Cohen CD, Hall MN, Ruegg MA, Inoki K, Walz G, Huber TB (2011) Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121:2197–2209
Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M (2010) Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology 51:1778–1788
Lee CH, Inoki K, Guan KL (2007) mTOR pathway as a target in tissue hypertrophy. Annu Rev Pharmacol Toxicol 47:443–467
Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H, Chen Y, Chen J (2007) Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol 27:495–502
Vogelbacher R, Wittmann S, Braun A, Daniel C, Hugo C (2007) The mTOR inhibitor everolimus induces proteinuria and renal deterioration in the remnant kidney model in the rat. Transplantation 84:1492–1499
Letavernier E, Legendre C (2008) mToR inhibitors-induced proteinuria: mechanisms, significance, and management. Transplant Rev (Orlando) 22:125–130
Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Ruegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL (2011) mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121:2181–2196
Fogo AB (2011) The targeted podocyte. J Clin Invest 121:2142–2145
Muller-Krebs S, Weber L, Tsobaneli J, Kihm LP, Reiser J, Zeier M, Schwenger V (2013) Cellular effects of everolimus and sirolimus on podocytes. PLoS One 8:e80340
Sartelet H, Toupance O, Lorenzato M, Fadel F, Noel LH, Lagonotte E, Birembaut P, Chanard J, Rieu P (2005) Sirolimus-induced thrombotic microangiopathy is associated with decreased expression of vascular endothelial growth factor in kidneys. Am J Transplant 5:2441–2447
Sandhu S, Wiebe N, Fried LF, Tonelli M (2006) Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 17:2006–2016
Douglas K, O’Malley PG, Jackson JL (2006) Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 145:117–124
Oda H, Keane WF (1999) Recent advances in statins and the kidney. Kidney Int Suppl 71:S2–5
Whaley-Connell A, Habibi J, Nistala R, Cooper SA, Karuparthi PR, Hayden MR, Rehmer N, DeMarco VG, Andresen BT, Wei Y, Ferrario C, Sowers JR (2008) Attenuation of NADPH oxidase activation and glomerular filtration barrier remodeling with statin treatment. Hypertension 51:474–480
Liu J, Zhang B, Chai Y, Xu Y, Xing C, Wang X (2015) Fluvastatin attenuated the effect of expression of beta1 integrin in PAN-treated podocytes by inhibiting reactive oxygen species. Mol Cell Biochem 398(1–2):207–215
Shibata S, Nagase M, Fujita T (2006) Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive rho signaling. J Am Soc Nephrol 17:754–764
Cormack-Aboud FC, Brinkkoetter PT, Pippin JW, Shankland SJ, Durvasula RV (2009) Rosuvastatin protects against podocyte apoptosis in vitro. Nephrol Dial Transplant 24:404–412
Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN, Lasaridis AN (2010) Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis 55:835–847
Kanjanabuch T, Ma LJ, Chen J, Pozzi A, Guan Y, Mundel P, Fogo AB (2007) PPAR-gamma agonist protects podocytes from injury. Kidney Int 71:1232–1239
Yang HC, Ma LJ, Ma J, Fogo AB (2006) Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int 69:1756–1764
Liu HF, Guo LQ, Huang YY, Chen K, Tao JL, Li SM, Chen XW (2010) Thiazolidinedione attenuate proteinuria and glomerulosclerosis in adriamycin-induced nephropathy rats via slit diaphragm protection. Nephrology (Carlton) 15:75–83
Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C, Ding G, Liu B, Yang T, Zhang A (2012) Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int 82:771–789
Agrawal S, Guess AJ, Benndorf R, Smoyer WE (2011) Comparison of direct action of thiazolidinediones and glucocorticoids on renal podocytes: protection from injury and molecular effects. Mol Pharmacol 80:389–399
Finlin BS, Bodles-Brakhop AM, Yao-Borengasser A, Zhu B, Starnes CP, McGehee RE Jr, Peterson CA, Kern PA, Rasouli N (2012) Regulation of small ubiquitin-like modifier-1, nuclear receptor coreceptor, histone deacetylase 3, and peroxisome proliferator-activated receptor-gamma in human adipose tissue. Metab Syndr Relat Disord 10:312–317
Simon M, Grone HJ, Johren O, Kullmer J, Plate KH, Risau W, Fuchs E (1995) Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol 268:F240–50
Muller-Deile J, Worthmann K, Saleem M, Tossidou I, Haller H, Schiffer M (2009) The balance of autocrine VEGF-A and VEGF-C determines podocyte survival. Am J Physiol Renal Physiol 297:F1656–67
Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE (2003) Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111:707–716
Wang H, Misaki T, Taupin V, Eguchi A, Ghosh P, Farquhar MG (2014) GIV/Girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J Am Soc Nephrol 26:314–327
Heydarian M, McCaffrey T, Florea L, Yang Z, Ross MM, Zhou W, Maynard SE (2009) Novel splice variants of sFlt1 are upregulated in preeclampsia. Placenta 30:250–255
Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111:649–658
Henao DE, Cadavid AP, Saleem MA (2013) Exogenous vascular endothelial growth factor supplementation can restore the podocyte barrier-forming capacity disrupted by sera of preeclamptic women. J Obstet Gynaecol Res 39:46–52
Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, Ponticelli C, Ritz E, Zucchelli P (1996) Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. the angiotensin-converting-enzyme inhibition in progressive renal insufficiency study group. N Engl J Med 334:939–945
Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, Lam A, O’Young G, Sannajust F, Stathis P, Schreiner G, Karumanchi SA, Protter AA, Pollitt NS (2007) Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension 50:686–692
Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, Shen W, Kopp JB, Thomas DB, Tufro A (2010) Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int 77:989–999
Sung SH, Ziyadeh FN, Wang A, Pyagay PE, Kanwar YS, Chen S (2006) Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol 17:3093–3104
de Vriese AS, Tilton RG, Elger M, Stephan CC, Kriz W, Lameire NH (2001) Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol 12:993–1000
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358:1129–1136
Müller-Deile J, Brocker V, Grunwald V (2010) Renal side effects of VEGF-blocking therapy. NDT Plus 3:172–175
Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K (2008) The notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med 14:290–298
Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP (1992) CD28-mediated signaling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 356:607–609
Walunas TL, Bakker CY, Bluestone JA (1996) CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med 183:2541–2550
Ishimoto T, Cara-Fuentes G, Wang H, Shimada M, Wasserfall CH, Winter WE, Rivard CJ, Araya CE, Saleem MA, Mathieson PW, Johnson RJ, Garin EH (2013) Serum from minimal change patients in relapse increases CD80 expression in cultured podocytes. Pediatr Nephrol 28:1803–1812
Garin EH, Reiser J, Cara-Fuentes G, Wei C, Matar D, Wang H, Alachkar N, Johnson RJ (2014) Case series: CTLA4-IgG1 therapy in minimal change disease and focal segmental glomerulosclerosis. Pediatr Nephrol 30:469–477
Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, Johnson RJ (2009) Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol 20:260–266
Yu CC, Fornoni A, Weins A, Hakroush S, Maiguel D, Sageshima J, Chen L, Ciancio G, Faridi MH, Behr D, Campbell KN, Chang JM, Chen HC, Oh J, Faul C, Arnaout MA, Fiorina P, Gupta V, Greka A, 3rd Burke GW, Mundel P (2013) Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med 369:2416–2423
Benigni A, Gagliardini E, Remuzzi G (2014) Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med 370:1261–1263
Alachkar N, Carter-Monroe N, Reiser J (2014) Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med 370:1263–1264
Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J (2008) Modification of kidney barrier function by the urokinase receptor. Nat Med 14:55–63
Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, Maiguel D, Karumanchi SA, Yap HK, Saleem M, Zhang Q, Nikolic B, Chaudhuri A, Daftarian P, Salido E, Torres A, Salifu M, Sarwal MM, Schaefer F, Morath C, Schwenger V, Zeier M, Gupta V, Roth D, Rastaldi MP, Burke G, Ruiz P, Reiser J (2011) Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med 17:952–960
Trimarchi H (2013) Primary focal and segmental glomerulosclerosis and soluble factor urokinase-type plasminogen activator receptor. World J Nephrol 2:103–110
Trimarchi H, Forrester M, Lombi F, Pomeranz V, Rana MS, Karl A, Andrews J (2014) Amiloride as an alternate adjuvant antiproteinuric agent in fabry disease: the potential roles of plasmin and uPAR. Case Rep Nephrol 2014:854521
Maas RJ, Deegens JK, Wetzels JF (2013) Serum suPAR in patients with FSGS: trash or treasure? Pediatr Nephrol 28:1041–1048
Cobos E, Jumper C, Lox C (2003) Pretreatment determination of the serum urokinase plasminogen activator and its soluble receptor in advanced small-cell lung cancer or non-small-cell lung cancer. Clin Appl Thromb Hemost 9:241–246
Edsfeldt A, Nitulescu M, Grufman H, Gronberg C, Persson A, Nilsson M, Persson M, Bjorkbacka H, Goncalves I (2012) Soluble urokinase plasminogen activator receptor is associated with inflammation in the vulnerable human atherosclerotic plaque. Stroke 43:3305–3312
Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR (2005) TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37:739–744
Greka A, Mundel P (2011) Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol 22:1969–1980
Krall P, Canales CP, Kairath P, Carmona-Mora P, Molina J, Carpio JD, Ruiz P, Mezzano SA, Li J, Wei C, Reiser J, Young JI, Walz K (2010) Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One 5:e12859
Heeringa SF, Moller CC, Du J, Yue L, Hinkes B, Chernin G, Vlangos CN, Hoyer PF, Reiser J, Hildebrandt F (2009) A novel TRPC6 mutation that causes childhood FSGS. PLoS One 4:e7771
Hauser PV, Pippin JW, Kaiser C, Krofft RD, Brinkkoetter PT, Hudkins KL, Kerjaschki D, Reiser J, Alpers CE, Shankland SJ (2010) Novel siRNA delivery system to target podocytes in vivo. PLoS One 5:e9463
Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, Bottinger EP (2001) Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 108:807–816
Drewe E, McDermott EM, Powell RJ (2000) Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 343:1044–1045
Akdogan MF, Gucun M, Denizli N, Guney M, Akdag I, Ozcan TB, Duranay M (2011) Complete reversal of nephrotic syndrome secondary to amyloidosis with use of infliximab in a patient with inflammatory bowel disease and ankylosing spondylitis. Ren Fail 33:531–533
Gottenberg JE, Merle-Vincent F, Bentaberry F, Allanore Y, Berenbaum F, Fautrel B, Combe B, Durbach A, Sibilia J, Dougados M, Mariette X (2003) Anti-tumor necrosis factor alpha therapy in fifteen patients with AA amyloidosis secondary to inflammatory arthritides: a followup report of tolerability and efficacy. Arthritis Rheum 48:2019–2024
Stokes MB, Foster K, Markowitz GS, Ebrahimi F, Hines W, Kaufman D, Moore B, Wolde D, D’Agati VD (2005) Development of glomerulonephritis during anti-TNF-alpha therapy for rheumatoid arthritis. Nephrol Dial Transplant 20:1400–1406
Chugh SS, Mace C, Clement LC, Del Nogal AM, Marshall CB (2014) Angiopoietin-like 4 based therapeutics for proteinuria and kidney disease. Front Pharmacol 5:23
Niethamer TK, Yardeni T, Leoyklang P, Ciccone C, Astiz-Martinez A, Jacobs K, Dorward HM, Zerfas PM, Gahl WA, Huizing M (2012) Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab 107:748–755
Noh H, King GL (2007) The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl 106:S49–53
Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, Meier M, Gueler F, Fiebeler A, Bahlmann FH, Leitges M, Haller H (2004) Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes 53:2101–2109
Ohshiro Y, Ma RC, Yasuda Y, Hiraoka-Yamamoto J, Clermont AC, Isshiki K, Yagi K, Arikawa E, Kern TS, King GL (2006) Reduction of diabetes-induced oxidative stress, fibrotic cytokine expression, and renal dysfunction in protein kinase Cbeta-null mice. Diabetes 55:3112–3120
Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW (2005) The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care 28:2686–2690
Sahali D, Pawlak A, Le Gouvello S, Lang P, Valanciute A, Remy P, Loirat C, Niaudet P, Bensman A, Guellaen G (2001) Transcriptional and post-transcriptional alterations of IkappaBalpha in active minimal-change nephrotic syndrome. J Am Soc Nephrol 12:1648–1658
Coppo R, Camilla R, Porcellini MG, Peruzzi L, Gianoglio B, Amore A, Dapra V, Loiacono E, Fonsato V, Dal Canton A, Esposito C, Esposito P, Tovo PA (2012) Saquinavir in steroid-dependent and -resistant nephrotic syndrome: a pilot study. Nephrol Dial Transplant 27:1902–1910
Conflict of interest
The authors declare no Conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Answers
1. b
2. d
3. e
4. c
5. c
Rights and permissions
About this article

Cite this article
Müller-Deile, J., Schiffer, M. Podocyte directed therapy of nephrotic syndrome—can we bring the inside out?. Pediatr Nephrol 31, 393–405 (2016). https://doi.org/10.1007/s00467-015-3116-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-015-3116-4