
Abstract
Inner ear physiology is reviewed with emphasis on features common to renal physiology. Genetic disorders in transporters/channels for chloride (ClC-K), bicarbonate (Cl-/HCO3- exchanger), protons (H+-ATPase), sodium (ENaC, NKKC1, NBC3, NHE3), potassium (KCNQ1/KCNE1, Kcc4), and water (AQP4) in the inner ear and their relation to the kidney are discussed. Based on data from human disorders (with or without mouse counterparts) and mouse models (without human counterparts) this article focuses on the involvement of these transporters/channels in hearing loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the inner ear and the renal tubules share several common transporters/channels, disturbances in the function of the inner ear and renal tubules may occur at the same time. The number of disturbed transporters/channels in both organs is increasing. Both pediatric nephrologists and otologists should be aware of this and that linkage of some clinical consequences is possible.
The aim of this review is to give an overview of combined renal and inner ear transporter/channel defects. First, basic information on inner ear anatomy and physiology is provided, followed by a description of the different genetic disorders in transporters/channels in the inner ear in relation to the kidney. The cochlear part of the inner ear is the main focus of this review. Additional genetic causes of inner ear diseases are well covered in recent reviews [1, 2, 3].
Inner ear physiology
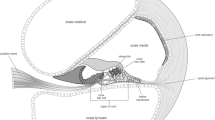
The inner ear or labyrinth is situated in the petrous part of the temporal bone. Within the otic capsule or bony labyrinth, lies the membranous labyrinth composed of the cochlea, vestibular system, and endolymphatic sac (Fig. 1). The major function of the inner ear is the transformation of mechanical stimuli (sound, motion, and gravitational force) into electrical signals that enable the perception of sound and the vestibular sensation. For this conversion, which occurs in the sensory hair cells of the cochlea (Fig. 1) (i.e., the organ of Corti; Fig. 2A) and the vestibular system (Fig. 1), the availability of K+ ions is essential.

Schematic anatomy of the various compartments of the membranous labyrinth. The inner ear spaces filled with endolymph are black and those filled with perilymph are white. [ B brain, C cochlea, CA cochlear aqueduct (not so widely patent as depicted here), CSF cerebrospinal fluid, DM dura mater, ES endolymphatic sac, ME middle ear, PB petrous bone, S sensory organ, SM scala media, ST scala tympani, SV scala vestibuli, V vestibular system]

A Cross-section of one turn of the cochlea depicting the organ of Corti ( C) composed of sensory hair cells and supporting cells , Claudius cells ( CC), external sulcus cells ( ESC), internal sulcus cells ( ISC), limbus spiralis ( Li), interdental cells ( IDC), Reissner’s membrane ( RM), and stria vascularis ( StV) adjacent to the fibrous spiral ligament ( SL) ( E endolymph, P perilymph). B High magnification of the stria vascularis composed of marginal cells (MC) facing the endolymph ( E), intermediate cells ( IC), and basal cells ( BC) adjacent to the spiral ligament ( SL) ( CAP capillary)
The central lumen (scala media) of the membranous labyrinth is filled with endolymph (Fig. 1, black area), a K+-rich/Na+-poor, electrically positively charged fluid with a neutral pH (Table 1) [4]. The surrounding spaces (scala tympani, scala vestibuli) within the bony labyrinth are filled with perilymph (Fig. 1, white area; Table 1) [4]. The perilymph of scala vestibuli and scala tympani differs in origin. It is assumed that the origin of the scala vestibuli perilymph is an ultrafiltrate from the capillary network in the inner ear. Tracer studies revealed that cerebrospinal fluid can account for 80% of the formation of the perilymph of the scala tympani via the cochlear aqueduct (Fig. 1). Perilymph rather than plasma should be considered as the precursor of the scala media endolymph. Homeostasis of the unique composition of the endolymph predominantly depends on secretion and/or absorption activities of specialized epithelial cells in the inner ear [5, 6].
It has become quite clear that in the inner ear the cochlear stria vascularis (Fig. 2A, B) plays an important role in endolymph homeostasis through large K+ conductance capacities, which result in the generation of a positive transepithelial potential of cochlear endolymph (i.e., the endocochlear potential) of +80 mV. The multilayered stria vascularis is composed of three cell types: marginal cells facing the endolymph, intermediate cells, and basal cells adjacent to the fibrous spiral ligament (Fig. 2B) [7].
Tight junctions of marginal and basal cells separate the endolymph from the perilymph-like fluid pervading the spiral ligament. The marginal cells, secreting K+ via apical K+ channels (KCNQ1/KCNE1 complex) into the endolymph, take up K+ basolaterally via distinct Na+K+-ATPases (α1β2 and α1β1) and Na+-K+-2Cl- (NKCC1) cotransporters [6, 8]. The localization of these and other transporters/channels in the marginal and intermediate cells are depicted in Fig. 3, which is a tentative but not complete scheme. Future research is aimed at the completion of this scheme. The basolaterally extruded Na+ by the marginal cells provides the driving force for Na+-K+-2Cl- cotransporters resulting in additional fluxes of K+ into the cell. The Cl- taken up is recycled back through basolateral electroneutral chloride channels (ClC-Ka/barttin and ClC-Kb/barttin) [9, 10]. Two sodium transporters localized in the apical membrane of the marginal cells, an amiloride-sensitive sodium channel (ENaC) and a sodium hydrogen exchanger (NHE3), are involved in the uptake of Na+ from the endolymph [11, 12]. Furthermore, an additional apical H+-ATPase is involved in the active secretion of H+ [13]. The basal and intermediate cells are coupled via gap junctions. In the intrastrial space between marginal and intermediate cells the potential is +80 mV realized through specific K+ channels (KCNJ10) in the intermediate cell membrane [14]. The marginal cell layer maintains a low K+ concentration in this space and apically secretes K+ driven by a difference in transmembrane potential between 0 and +10 mV. The molecular mechanisms that generate the endocochlear potential have recently been reviewed by Wangemann [15].

Schematic localization of transporters/channels in the cochlear stria vascularis, i.e., in the marginal ( MC) and intermediate cells ( IC) ( IS intrastrial space)
K+ flows from the endolymph through apical transduction channels that are opened by movement of sensory hair cells and leaves these cells via K+ channels (e.g., KCNQ4). From the vicinity of the outer hair cells, K+ diffuses back via gap junctions formed by connexins into the spiral ligament and further towards the stria vascularis and is finally released into the endolymph. The most significant recycling pathways of K+ ions in the cochlea are illustrated in Fig. 4. From the inner hair cells K+ is recycled back into the endolymph via internal sulcus cells or via spiral limbus fibrocytes and interdental cells. In addition external sulcus cells are also involved in the recycling of K+ [5, 6]. Knowledge of the components involved in the pathways for K+ recycling other than through the marginal cells is far from complete and needs to be pursued further.

Schematic representation of a cochlear turn with the most-significant recycling pathways of K+ ions illustrated by arrows. Furthermore it depicts the organ of Corti composed of sensory inner ( IHC) and outer ( OHC) hair cells and supporting cells [i.e., inner ( IPC) and outer pillar ( OPC) cells, Deiters cells ( DC), and Hensen cells ( HC)], Claudius cells ( CC), external sulcus cells ( ESC), internal sulcus cells ( ISC), spiral limbus ( Li), interdental cells ( IDC), Reissner’s membrane ( RM), and stria vascularis ( StV) adjacent to the fibrous spiral ligament ( SL) (modified with permission of Professor G. van Camp, University of Antwerp, Belgium)
Transport disorders
The composition of the endolymphatic fluid that bathes the sensory hair cells of Corti’s organ (Fig. 2A) is strictly regulated to enable the transduction of sound into neural signals. Abnormalities in volume or composition of the endolymph will adversely affect hearing. Mutations in transporters or channels common to inner ear and/or kidney in humans and/or mouse models are presented in brief (Table 2).
Human disorders (with or without mouse counterparts)
Chloride channels in Bartter syndromes
Bartter syndrome is clinically characterized by salt wasting, hypokalemic alkalosis, hyperreninemia, hyperaldosteronism with hyperplasia of the juxtaglomerular apparatus, and sensorineural hearing loss in some subtypes. Bartter syndrome types I through IV (autosomal recessive disorders), in which the thick ascending limb of Henle’s loop is affected, result from mutations in genes encoding for different transporters/channels (schematically depicted in Fig. 5). Not only type K chloride channels (ClC-K, Bartter syndrome types III and IV) may be affected but also the Na+-K+-2Cl- cotransporter (NKCC2) and the ROMK2 potassium channel, causing Bartter syndrome type I and II, respectively. In contrast to ClC-K, the NKKC2 and ROMK2 are not detected in the cochlea. Functional ClC-K is composed of two subunits, i.e., barttin protein is associated with either human ClC-Ka or ClC-Kb (rodent orthologues ClC-K1 and ClC-K2, respectively). Enhanced expression of barttin (associated with either ClC-K1 or ClC-K2) studied in Xenopus laevis oocytes resulted in an increase in current amplitude, a difference in calcium sensitivity, and an enhanced ClC-K abundance in the cell membrane [16]. In the cochlea ClC-K1, ClC-K2 as well as barttin, is found in the basolateral membrane of marginal cells (Fig. 3) [10]. Mutations in the gene encoding the protein barttin ( BSND) affect both chloride channels (ClC-Ka/barttin and ClC-Kb/barttin), causing the sensorineural hearing loss observed in Bartter syndrome type IV, while a defect in ClC-Kb alone (Bartter syndrome type III) does not cause deafness as ClC-Ka is still functioning. Knock-out of ClC-K1 ( Clcnk1-/-), which is present in the thin ascending limb of Henle, leads to nephrogenic diabetes insipidus in mice [17]. In the absence of this chloride channel, a lower osmolality is observed in the inner medulla of mice, although no defect in humans has been observed yet. A remarkable digenic defect has recently been described by Schlingmann et al. [18]. They determined mutations in the genes encoding ClC-Ka ( CLCNKA) as well as ClC-Kb ( CLCNKB) within one patient. This patient displayed similar clinical symptoms to those seen in Bartter syndrome type IV.

Schematic localization of the transporters/channels in renal thick ascending limb cells ( TALC) that can be affected in Bartter syndrome types I through IV, i.e., NKCC2 / type I, ROMK2 / type II, ClC-Kb / type III, and barttin / type IV ( I interstitium, L luminal)
Chloride-bicarbonate exchangers in Pendred syndrome
Pendred syndrome, in which a mutation affects the pendrin gene SLC26A4 (PDS), is an autosomal recessive disorder characterized by goiter and sensorineural deafness. The pendrin gene has a relatively restricted pattern of expression in the kidney, thyroid, and inner ear. In the kidney pendrin functions as an apically located Cl-/HCO3- exchanger present in the type B intercalated cells of the distal nephron. In these cells H+ is secreted into the interstitium, while HCO3- exits across the apical membrane into the lumen (Fig. 6) [19]. However, patients with Pendred syndrome do not display metabolic alkalosis [20]. In distinct thyroid follicular cells, pendrin is localized in the apical membrane where it functions as a Cl-/I- exchanger. In patients with Pendred syndrome impairment of this exchanger leads to a disturbed iodide organification as determined by a positive perchlorate discharge test [21]. In the inner ear pendrin gene expression is detected in distinct areas that are putatively critical in the regulation of endolymphatic fluid composition such as cochlear external sulcus cells (Fig. 2A), vestibular transitional cells, and endolymphatic sac cells (Fig. 1) [22]. Examination of the embryogenesis of pendrin knock-out mice ( Pds -/-) suggests a disturbance in endolymph resorption, resulting in an increased endolymphatic fluid volume at a critical developmental stage [23]. However, the exact role of pendrin in the inner ear remains to be defined.

Schematic localization of the transporters involved in acid/base homeostasis in renal A ( A IC) and B ( B IC) intercalated cells
Proton pumps in renal tubular acidosis and sensorineural hearing loss
Renal tubular acidosis is a clinical syndrome in which an inherited renal tubular defect leads to a failure in secretion of H+ and maintenance of a normal plasma HCO3- concentration (metabolic acidosis). The proton pump H+-ATPase consists of two functional domains: the membrane-spanning V0 domain responsible for proton translocation and the V1 domain, composed of a hexamer of three A-subunits and three B-subunits and at least six other different subunits, functioning in ATP hydrolysis. Apically located renal proton pumps display tissue-specific isoforms, i.e., B1 of the V1 domain and α4 of the V0 domain. The genes encoding B1 ( ATP6V1B1) and α4 ( ATP6V0A4) are affected in the autosomal recessive form of distal renal tubular acidosis, which is characterized by a disturbance in the function of H+-ATPase present at the apical surface of type A intercalated cells of the distal nephron [24, 25]. In these cells luminal H+ secretion is linked to basolateral exchange of Cl- and HCO3- (Fig. 6). Patients with a B1 mutation display sensorineural hearing loss with childhood onset in addition to distal renal tubular acidosis. During longer follow-up, it has become evident that mild and/or later-onset hearing loss can also be present in some patients with the α4 mutation [26]. In the cochlea the strongest immunoreactivity for H+-ATPase is detected in the apical membrane and cytoplasm of marginal cells (Fig. 3). Apical staining is also observed in cochlear interdental cells (Fig. 2A) and endolymphatic sac cells (Fig. 1) [13, 25]. In the normal inner ear cochlear endolymph pH is maintained at 7.4, while in the endolymphatic sac it is 6.6. A fine regulation of the cochlear endolymph pH is apparently a prerequisite for adequate hearing. Remarkably mice lacking the B1-subunit of H+-ATPase ( Atp6b1-/-) display a normal hearing [27].
Sodium channels
Mutations in transmembrane serine protease (TMPRSS3), an activator of the epithelial sodium channel (ENaC), are observed in both familial and sporadic cases of autosomal recessive sensorineural deafness, i.e., DFNB8/10 [28, 29]. Studies in Xenopus laevis oocytes demonstrated that, after auto-catalytic processing, TMPRSS3 activates ENaC. In the cochlea ENaC is located apically in the marginal cells (Fig. 3) and plays a role in maintaining a low Na+ concentration in the endolymph [11]. Mutations in the α, β, and γ subunits of ENaCs are found in patients with autosomal recessive pseudo-hypoaldosteronism type I [28]. This disease is characterized by tubular unresponsiveness to the action of aldosterone as manifested by salt-wasting and hyperkalemia. Studies of subtle hearing loss in this patient population are currently underway.
Potassium channels in Jervell and Lange-Nielsen syndrome
Long QT syndrome is a disorder with ventricular repolarization and a prolonged QT interval. This may result in syncope, seizures, and sudden death due to cardiac arrhythmias. The autosomal recessive form is the Jervell and Lange-Nielsen syndrome (JLNS), which is associated with profound bilateral sensorineural deafness [30]. Knock-out mice for KCNQ1 or KCNE1 encompass mouse models for JLNS ( Kcnq1-/- and Kcne1 -/-) [31, 32]. Mutation in either gene is associated with JLNS in humans [33]. KCNE1 is essential for KCNQ1 membrane targeting and/or stability of KCNQ1 in the membrane. KCNE1 co-assembles with KCNQ1 and both are localized in the apical membrane of cochlear marginal cells (Fig. 3) and vestibular dark cells, realizing K+ secretion into the endolymph [6]. In KCNE1 knock-out mice the proximal tubular function is disturbed, since the KCNQ1/KCNE1 complex is a luminal K+ channel in proximal tubular cells, which generates the driving force for electrogenic Na+ and substrate (glucose) reabsorption [32]. No information is yet available about the function of the proximal tubule in JLNS in humans.
Mouse models (without human counterparts)
Sodium-potassium-chloride cotransporters
The secretory Na+-K+-2Cl- cotransporter (isoform NKCC1) is localized in the basolateral membrane of cochlear marginal cells (Fig. 3) and is involved in the influx of Na+, K+, and Cl− [6]. The other absorptive isoform NKKC2 is exclusively expressed in the kidney in the thick ascending limb of Henle (Fig. 5; affected in Bartter syndrome type I). Both isoforms display a difference in transcript size and share a relatively low overall amino acid homology and are products of different genes [34]. The absence of NKCC1 in knock-out mice ( Slc12a2-/-) results in a drastic decrease in secretion of K+ into the endolymph and the collapse of the cochlear Reissner’s membrane. The supply of Na+ (effluxed by Na+K+-ATPase of the marginal cells; Fig. 3) is interrupted, consequently limiting the uptake of K+. Both hearing and balance are affected in these mice [35]. In humans reversible hearing loss is observed after administration of high doses of furosemide, which inhibits NKKC1, temporarily disrupting the endocochlear potential [36]. Although NKCC1 is present in the basolateral membrane of the outer medullary collecting duct cells in the rat, its physiological role remains to be determined [37].
Sodium-bicarbonate cotransporters
The Na+-HCO3- cotransporter NBC3 plays an important role in cochlear spiral ligament fibrocyte sodium bicarbonate transport, and subsequently preserves the function of the sensory inner and outer hair cells of Corti’s organ (Fig. 2A) [38]. In the kidney NBC3 is localized in the collecting duct where it may modulate the activity of H+-ATPase [39]. However, loss of NBC3 in knock-out mice ( Slc4a7-/-) causes hearing loss and blindness only due to the degeneration of sensory receptors. This indicates that the loss of NBC3 in the inner ear and eye cannot be compensated by other mechanisms in contrast to the kidney [38].
Sodium-hydrogen exchangers
Expression of the Na+/H+ exchanger type 3 (NHE3) in the cochlea is limited to the apical surface of marginal cells (Fig. 3) [12]. Impaired renal acid/base balance and disturbed Na+ fluid volume homeostasis is observed in NHE3 knock-out mice ( Slc9a3-/-) [40]. It remains to be established whether this is associated with hearing problems.
Potassium-chloride cotransporters
K+-Cl- cotransporter Kcc4 in the cochlea is restricted to the supporting cells (Deiters cells) of the sensory outer hair cells of Corti’s organ and plays a role in siphoning K+, after its exit from these cells (Fig. 2A). Knock-out mice for Kcc4 ( Slc12a7-/-) display impaired hearing and distal renal tubular acidosis. The distal renal tubular acidosis is supposed to be due to an impairment of Cl- recycling across the basolateral membrane of type A intercalated cells of the distal nephron (Fig. 6). The hearing loss may be a result of the disturbed K+ integrity around the outer hair cells [41].
Water channels
Impaired hearing and a mild urinary concentrating defect is observed in knock-out mice lacking aquaporin 4 water channels ( Aqp4-/-). Aquaporin 4 is responsible for a substantial amount of the basolateral water movement in the inner medulla of the kidney. In the cochlea aquaporin 4 is expressed in the inner sulcus cells, supporting cells (Hensen cells) of the organ of Corti and in Claudius cells (Fig. 2A). It is assumed that aquaporin 4 facilitates rapid osmotic equilibration in cochlear cell types that are subject to large K+ fluxes during signal transduction [42].
Miscellaneous
Mitochondrial diseases
Special attention should be given to the analysis of mitochondrial DNA because progressive hearing loss is frequently observed in patients with mitochondrial disease [43]. In the inner ear mitochondrial ATP production is required for the active secretion of H+ and K+. The progressive nature of this hearing loss can probably be explained by heteroplasmy. When both normal and mutant mitochondrial DNA is present in mother cells, some daughter cells may drift towards more mutant DNA. The most common renal manifestations of mitochondrial disorders are Fanconi syndrome, focal glomerular sclerosis, and progressive tubulointerstitial nephropathy. Thus, these clinical manifestations in combination with hearing loss necessitate mitochondrial DNA screening.
Calcium/magnesium transporters/channels
Regulation of Ca2+ and Mg2+ transport in the endolymph is still an open area; the cochlear endolymph concentrations are extremely low with Ca2+ being 0.02 and Mg2+ 0.01 mmol/l (Table 1). However, calcium is required for an adequate functioning of sensory hair cells. A mutation in the gene encoding Ca2+-ATPase, localized in the sensory hair cells, causes deafness and vestibular/motor disturbances in mice [44].
Water channels
Another interesting observation that needs to be further investigated is the extensive expression of aquaporin 2 in the epithelial lining of the cochlear Reissner’s membrane [45] in relation to the difference in osmolarity between scala media endolymph and scala vestibuli perilymph (Table 1) [46]. Children with a defective aquaporin 2 system (nephrogenic diabetes insipidus) apparently display normal hearing.
Future perspectives
It is expected that the genetic basis for most if not all hereditary disorders causing hearing loss will eventually be known. Gene expression profiles through cDNA microarray analysis of inner ear tissues are a new promising tool [47, 48]. This will allow better genetic counseling.
Is there a prospective treatment of hereditary disorders causing hearing loss? In tubular transport disorders of the kidney, a defect in one segment of the tubule may sometimes be compensated by altered activity in other segments of the tubule. For example, amiloride treatment in Bartter syndrome type I, with a defect in the thick ascending limb of the loop of Henle, can considerably decrease the K+ loss in the distal tubule and collecting duct. Elucidation of compensatory transport mechanisms in the inner ear (in particular in the endolymphatic sac [49, 50, 51]) may offer some new perspectives. The inner ear is also an attractive target for gene therapy, because the transgenes may be delivered locally [52, 53].
Synergy of research combining otological and nephrological expertise is a promising area. Within inner ear research it is expected that the increase in knowledge of electrolyte transport mechanisms and water movements will provide a more complete overview of the structures and processes involved in endolymph homeostasis, which may ultimately lead to the development of new preventive and therapeutic strategies for hereditary disorders causing hearing loss.
References
Willems PJ (2000) Genetic causes of hearing loss. N Engl J Med 342:1101–1109
Tekin M, Arnos KS, Pandya A (2001) Advances in hereditary deafness. Lancet 358:1082–1090
Couloigner V, Sterkers O, Friedlander G, Ferrary E (2003) Syndromes ‘reins-oreilles’: aspects moléculaires. Actualités Néphrolologiques Flammarion, Paris, pp 147–161
Ferrary E, Sterkers O (1998) Mechanisms of endolymph secretion. Kidney Int 65:98–103
Salt AN (2001) Dynamics of the inner ear fluids. In: Jahn AF, Santos-Sacchi J (eds) Physiology of the ear. Singular, San Diego, pp 333–355
Wangemann P (2002) K+ cycling and its regulation in the cochlea and the vestibular labyrinth. Audiol Neurootol 7:199–205
Peters TA. Kuijpers W, Tonnaer ELGM, Muijen van GNP, Jap PHK (1995) Distribution and features of melanocytes during inner ear development in pigmented and albino rats. Hear Res 85:169–180
Peters TA, Kuijpers W, Curfs JHAJ (2001) Occurrence of NaK-ATPase isoforms during rat inner ear development and functional implications. Eur Arch Otorhinolaryngol 258:67–73
Sage CL, Marcus DC (2001) Immunolocalization of ClC-K chloride channel in strial marginal cells and vestibular dark cells. Hear Res 160:1–9
Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, Jentsch TJ (2001) Barttin is a Cl- channel β subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 414:558–561
Couloigner V, Fay M, Djelidi S, Farman N, Escoubet B, Runembert I, Sterkers O, Friedlander G, Ferrary E (2001) Location and function of the epithelial Na channel in the cochlea. Am J Physiol Renal Physiol 280:F214–F222
Bond BR, Ng LL, Schulte BA (1998) Identification of mRNA transcripts and immunohistochemical localization of Na/H exchanger isoforms in gerbil inner ear. Hear Res 123:1–9
Stankovic KM, Brown D, Alper SL, Adams JC (1997) Localization of pH regulating proteins H+-ATPase and Cl-/HCO3- exchanger in the guinea pig inner ear. Hear Res 114:21–34
Marcus DC, Wu T, Wangemann P, Kofuji P (2002) KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 282:C403–C407
Wangemann P (2002) K+ cycling and the endocochlear potential. Hear Res 165:1–9
Waldegger S, Jeck N, Bart P, Peters M, Vizthum H, Wolf K, Kurtz A, Konrad M, Seyberth HW (2002) Barttin increases surface expression and changes current properties in ClC-K channels. Pflug Arch Eur J Physiol 444:411–418
Uchida S, Maromo F (2000) Severity impaired urine-concentrating ability in mice lacking the ClC-K1 chloride channel. Exp Nephrol 8:301–305
Schlingmann KP, Konrad M, Jeck N, Waldegger P, Reinalter SC, Holder M, Seyberth HW, Waldegger S (2004) Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 350:1314–1319
Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW (2003) Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol 284:F229–F241
Petrovic S, Wang Z, Ma L, Soleimani M (2003) Regulation of the apical Cl-/HCO3- exchanger pendrin in rat collecting duct in metabolic acidosis. Am J Physiol Renal Physiol 284:F103–F112
Royaux I, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED (2000) Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141:839–845
Everett LA, Morsli H, Wu DK, Green ED (1999) Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggest a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A 96:9727–9732
Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10:153–161
Karet FE (2002) Inherited distal renal tubular acidosis. J Am Soc Nephrol 13:2178–2184
Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CWRJ, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skovorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21:84–90
Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch ABS, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez-Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE (2002) Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 39:796–803
Dou H, Friberg K, Cardell EL, Lifton R, Choo D (2003) Mice lacking the B1 subunit of H+-ATPase have normal hearing. Hear Res 180:76–84
Guipponi M, Vuagniaux G, Wattenhofer M, Shibuya K, Vazquez M, Dougherty L, Scamuffa N, Guida E, Okui M, Rossie C, Hancock M, Buchet K, Reymond A, Hummler E, Marzella PL, Kudoh J, Shimizu N, Scott HS, Antonarakis SE, Rossier BC (2002) The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet 11:2829–2836
Lee YJ, Park D, Kim SY, Park WJ (2003) Pathogenic mutations but no polymorphisms in congenital and childhood onset autosomal recessive deafness disrupt the proteolytic activity of TMPRSS3. J Med Genet 40:629–631
Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15:186–189
Casimiro MC, Knollmann BV, Ebert SN, Vary JC Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K (2001) Targeted disruption of the kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen syndrome. Proc Natl Acad Sci U S A 98:2526–2531
Warth R, Barhanin J (2002) The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am J Physiol Regul Integr Comp Physiol 282:R639–R648
Tyson J, Tranebjaerg L, Bellman S, Wren C, Taylor JFN, Bathen J, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M (1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet 6:2179–2185
Russell JM (2000) Sodium-potassium-chloride cotransport. Physiol Rev 80:211–276
Delpire E, Lu J, England R, Dull C, Thorne T (1999) Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22:192–195
Rybak LP (1993) Ototoxicity of loop diuretics. Otolaryngol Clin North Am 26:829–844
Wall SM, Fischer MP, Metha P, Hassell KA, Park SJ (2001) Contribution of the Na+-K+-2Cl- cotransporter NKCC1 to Cl- secretion in rat OMCD. Am J Physiol Renal Physiol 280:F913–F921
Bok D, Galbraith G, Lopez, Woodruff M, Nusinowitz S, BeltrandelRio H, Huang W, Zhao S, Geske R, Montgomery C, Sligtenhorst I van, Friddle C, Platt K, Sparks MJ, Pushkin A, Abuladze N, Ishiyama A, Dukkipati R, Liu W, Kurtz I (2003) Blindness and auditory impairment caused by loss of the sodium bicarbonate cotransporter NBC3. Nat Genet 34:313–319
Pushkin A, Yip KP, Clark I, Abuladze N, Kwon TH, Tsuruoka S, Schwartz GJ, Nielsen S, Kurtz I (1999) NBC3 expression in rabbit collecting duct; colocalization with vacuolar H+-ATPase. Am J Physiol Renal Physiol 46:F974–F981
Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull G (1998) Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet 19:282–285
Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416:874–878
Li J, Verkman AS (2001) Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem 278:31233–31237
Rotig A, Munnich A (2003) Genetic features of mitochondrial respiratory chain disorders. J Am Soc Nephrol 14:2995–3007
Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K (1998) Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat Genet 19:390–394
Mhatre AN, Jero J, Chiappini I, Bolasco G, Barbara M, Lalwani AK (2002) Aquaporin-2 expression in the mammalian cochlea and investigation of its role in Meniere’s disease. Hear Res 170:59–69
Sterkers O, Ferrary E, Amiel C (1984) Inter-and intracompartmental osmotic gradients within the rat cochlea. Am J Physiol 247:F602–F606
Lin J, Ozeki M, Javel E, Zhao Z, Pan W, Schlentz E, Levine S (2003) Identification of gene expression profiles in rat ears with cDNA microarrays. Hear Res 175:2–13
Abe S, Katagiri T, Saito-Hisaminato A (2003) Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet 72:73–82
Peters TA, Tonnaer ELGM, Kuijpers W, Cremers CWRJ, Curfs JHAJ (2002) Differences in endolymphatic sac mitochondria-rich cells indicate specific functions. Laryngoscope 112:534–541
Peters TA, Tonnaer ELGM, Kuijpers W, Curfs JHAJ (2003) Changes in ultrastructural characteristics of endolymphatic sac ribosome-rich cells of the rat during development. Hear Res 176:94–104
Couloigner V, Teixeira M, Sterkers O, Rask-Andersen H, Ferrary E (2004) The endolymphatic sac: its role in the inner ear. Med Sci (Paris) 20:304–310
Ishimoto S, Kawamoto K, Kanzaki S, Raphael Y (2002) Gene transfer into supporting cells of the organ of Corti. Hear Res 173:187–197
Han D, Yu Z, Fan E, Liu C, Liu S, Li Y, Liu Z (2004) Morphology of auditory hair cells in guinea pig cochlea after transgene expression. Hear Res 190:25–30
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peters, T.A., Monnens, L.A.H., Cremers, C.W.R.J. et al. Genetic disorders of transporters/channels in the inner ear and their relation to the kidney. Pediatr Nephrol 19, 1194–1201 (2004). https://doi.org/10.1007/s00467-004-1626-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-004-1626-6