
Abstract
Purpose
Glioblastoma multiforme is the most frequent primary brain tumor, it has poor prognosis, and it remains refractory to current treatment. The success of temozolomide (TMZ) appears to be limited by the occurrence of chemoresistance. Recently, we report the use of pertussis toxin as adjuvant immunotherapy in a C6 glioma model; showing a decrease in tumoral size, it induced selective cell death in Treg cells, and it elicited less infiltration of tumoral macrophages. Here, we evaluated the cytotoxic effect of pertussis toxin in combination with TMZ for glioma treatment, both in vitro and in vivo RG2 glioma model.
Methods
We determined cell viability, cell cycle, apoptosis, and autophagy on treated RG2 cells through flow cytometry, immunofluorescence, and Western blot assays. Twenty-eight rats were divided in four groups (n = 7) for each treatment. After intracranial implantation of RG2 cells, animals were treated with TMZ (10 mg/Kg/200 μl of apple juice), PTx (2 μg/200 μl of saline solution), and TMZ + PTx. Animals without treatment were considered as control.
Results
We found an induction of apoptosis in around 20 % of RG2 cells, in both single treatments and in their combination. Also, we determined the presence of autophagy vesicles, without any modifications in the cell cycle in the TMZ – PTx-treated groups. The survival analyses showed an increase due to individual treatments; while in the group treated with the combination TMZ − PTx, this effect was enhanced.
Conclusion
We show that the concomitant use of pertussis toxin plus TMZ could represent an advantage to improve the glioma treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is the most common cause of death in many countries. Worldwide, central nervous system (CNS) tumors are an important cause of morbidity and mortality. It was estimated that approximately 30 % of all primary and 80 % of all malignant brain tumors are accounted for by the broad category of gliomas, while 54 % of all malignant brain tumors are glioblastoma multiforme (GBM) and they occur at a rate of 3.2 per 100,000 person per year (National Cancer Institute 2011; Deorah et al. 2006). At the Instituto Nacional de Neurología y Neurocirugía of Mexico, GBM represents 9 % of all brain tumors and the 45.7 % of primary gliomas (Velasquez-Perez and Jimenez-Marcial 2003; Lopez-Gonzalez and Sotelo 2000). GBM comprises a heterogeneous group of neoplasms that differ in their location within the CNS, age and gender distribution, growth potential, extent of invasiveness, morphological features, tendency for progression, and response to treatments.
Although several innovative therapies are being developed for GBM, the mainstays of conventional treatment combine surgical resection with radiation and an oral DNA alkylating agent, temozolomide (TMZ); the overall median survival does not extend beyond 14 months (Stupp et al. 2005, 2007; Buckner 2003). TMZ is considered as a standard chemotherapeutic regimen for the first-line treatment for malignant gliomas (Ryu et al. 2012). TMZ exerts its cytotoxics effects mainly through the induction of apoptosis. It interferes with methylation process, and it is known that the persistence of some methylated guanine residues in DNA is necessary for the triggering of the programmed cell death process (Denny et al. 1994), However, TMZ does not exhibit uniform sensitivity to malignant gliomas, and its survival benefit remains unsatisfactory, mainly because the success of TMZ in GBM tumors appears to be limited by the occurrence of chemo resistance that allows glioma cells to escape from death signaling pathways (Carmo et al. 2011; Ryu et al. 2012).
Due to a lack of response to treatment in gliomas, some of the current challenges may be overcome by the integration of novel study designs, the selective use of targeted agents depending on tumor molecular characteristics, and simultaneous inhibition of several survival cellular pathways. Pertussis toxin (PTx), an exotoxin produced by Bordetella pertussis, regulates the activation induced of apoptotic cell death and autophagic process (Carracedo et al. 1995; Ogier-Denis and Codogno 2003). Previously, we reported the potential use of PTx as an immunomodulatory molecule on glial tumors induced by C6 glioma cells, demonstrating the selective induction of apoptosis on Treg cells with decrements in C6 cell viability and the increment of apoptosis in glioma cells. Also, PTx had an effect blocking the migration of glioma invading macrophages (Orozco-Morales et al. 2012). Therefore, the aim of this study was to determine the cytotoxic effect of PTx treatment in combination with TMZ as adjuvant therapy both in vivo and in vitro assays in RG2 glioma model.
Materials and methods
Reagents
Temozolomide (TMZ, 3,4-dihydro-3-methyl-4-oxoimidazo [5,1-d]-as-tetrazine-8-carboxamide) was purchased from Sigma-Aldrich. It was dissolved in DMSO (Sigma Chemical Co., St. Louis, MO, USA) for assays. Acridine orange was obtained from Polysciences (Warrington, PA, USA). Lyophilized powder of PTx was purchased from Sigma-Aldrich (Sigma Chemical Co., St. Louis, MO, USA), and it was dissolved in saline solution.
Cell culture
Tumor cell line RG2 was purchased from ATCC (American Tissue Culture Collection, Rockville, MD, USA). Cells were cultured under sterile conditions at 37 °C in a humid environment with 5 % of CO2 in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO BRL, Grand Island, NY, USA) supplemented with 10 % fetal bovine serum (GIBCO BRL), 4 mM glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. Cultures were regularly checked and split when the cell confluence reached 85 %.
In vitro assays
Cells were seeded at a density of 3 × 105 cells/well in 6-well Costar plates precoated with poly-l-lysine (5 μg/ml) and incubated overnight at 37 °C. After exposure to DMSO (1 μl/ml) for control group, TMZ (100 μM), PTx (20 ng/ml), or TMZ + PTx for 48 h, the cells were trypsinized and collected for apoptosis detection (Annexin V/7AAD), LC3 detection by immunofluorescence, immunoblotting, cell cycle, and acridine orange vesicles analysis. The number of living cells was estimated by trypan blue exclusion. To inhibit autophagy, we used 3-methyladenine (3-MA), an inhibitor of phosphatidylinositol 3-kinase (PI3K), which inhibits autophagic sequestration (Kabeya et al. 2000; Mizushima et al. 2001; Kim and Klionsky 2000). According to this, experimental groups were previously exposed for 24 h to DMSO 1 μl/ml (Control), TMZ (100 μM), PTx (20 and 200 ng/ml), or TMZ + PTx, respectively, as previously was described, and then, 1.0 mM of 3-MA was added for additional 48 h. Cells were monitored under light microscope and later examined by immunofluorescence to detect LC3 protein.
Detection of acidic vesicular organelles
Autophagy is characterized by the formation and promotion of acidic vesicular organelles (AVOs) (Kanzawa et al. 2003a). We take advantage of the lysosomotropic agent acridine orange that moves freely across biological membranes when it is uncharged. Its protonated form accumulates in acidic cell compartments, where it forms aggregates that fluorescence bright red (Paglin et al. 2001; Kanzawa et al. 2003b). Flow cytometry with acridine orange staining was employed to detect and quantify the AVOs. In acridine orange-stained cells, the cytoplasm and nucleus fluoresce bright green and dim red, respectively, whereas acidic compartments fluoresce bright red, as it is described elsewhere (Kanzawa et al. 2003a). Therefore, we could measure a change in the intensity of the red fluorescence to represent the percentage of their cellular acidic compartment. After treatment with TMZ alone or TMZ combined with PTx, 1 × 105 cells were collected in phenol red-free RPMI 1640 medium. The green (FL-1) and red (FL-3) fluorescence of acridine orange was measured with a flow cytometer using the CellQuest software (Becton–Dickinson, San Jose, CA, USA). The sum of the upper-left and the upper-right quadrants of the dot plot was used to represent the percentage of autophagy. These assays were done by triplicate.
LC3 and Beclin immunofluorescence staining
The microtubule-associated protein 1 light-chain 3 (LC3) is essential for amino-acid starvation-induced autophagy, and it is associated with the autophagosome membrane (Kabeya et al. 2000) (Munafo and Colombo 2001). Tumor cells were cultured on chamber slide dishes (BD Biosciences, Bedford, MA, USA). Once the treatments previously mentioned were assayed, cells were fixed in 4 % paraformaldehyde, blocked with 3 % normal goat serum, and then incubated in 1 % BSA/10 % normal goat serum/0.3 M glycine in 0.1 % PBS-Tween for 1 h to permeabilize the cells and to block non-specific protein–protein interactions. After that, cells were incubated either with the goat polyclonal anti-LC3A/B or with the rabbit polyclonal anti-Beclin 1 antibody (abcam, Cambridge, UK) for 30 min. Then, cells were washed twice with PBS and incubated by additional 30 min in darkness with an anti-goat IgG antibody or anti-rabbit IgG antibody (abcam, Cambridge, UK), washed again with PBS, and finally mounted with mounting fluid. Images were obtained on a Leica microscope.
Apoptosis detection with Annexin V assay and PI double stain
After the experimental treatments described above were administered, cells were rinsed with PBS and resuspended in 1 ml of filtered PBS and adjusted to a final concentration of 1 × 106 cells/ml. Cell suspensions were incubated with APC-labeled Annexin V (Annexin V Apoptosis Detection Kit I, BD Pharmingen) and propidium iodide (PI) in 100 μl of binding buffer. After gently vortexing, cells were incubated for 15 min at room temperature in darkness and 400 μl of binding buffer was added and analyzed by flow cytometry within 1 h after treatments. A total of 10,000 events were evaluated. Data were collected on a FACSCalibur instrument (BD Biosciences). Cell QuestPro and Flow Jo ver. 7.6.1 software were used for data analysis.
The dot plot of the four quadrants in the figures was used to distinguish the normal (annexin V−/PI−), early apoptotic (annexin V+/PI−), late apoptotic (annexin V+/PI+), and necrotic (annexin V−/PI+) cells (Pietra et al. 2001).
Determination of cell cycle
The cell distribution in the cell cycle was determined by flow cytometry using PI staining. After experimental treatments, 1 × 106 cells were rinsed with PBS, collected, fixed in cold 70 % ethanol, and stored at −20 °C until analysis. Cells were washed with PBS, centrifuged and resuspended in permeabilization buffer (0.2 M Na2HPO4, 0.1 M acetic acid), and incubated 30 min at 37 °C. Then, cells were centrifugated and resuspended in RNase (100 μg/ml) and PI (25 μg ml−1). Cells were incubated in the dark for 30 min at room temperature. Cell cycle was determined on a FACSCalibur Flow cytometer FL-4 (Becton–Dickinson, USA). Measurements were obtained at 488 nm gating out doublets and clumps for each sample. A total of 10,000 events were evaluated. Cell QuestPro and Flow Jo ver. 7.6.1 software were used for data analysis.
Immunoblotting
Cells were disrupted with lysis buffer (25 mM Hepes, 1.5 % Triton X-100, 0.1 % SDS, 0.5 M NaCl, 5 mM EDTA, and 0.1 mM sodium deoxycholate) containing a protease inhibitor cocktail (Wang et al. 2008). Total cellular extracts were analyzed using the Bio-Rad protein assay dye reagent, and an equal amount of proteins from each group was separated using SDS–PAGE, followed by transfer to PVDF membranes. Membranes were incubated with 5 % skim milk solution (blocking solution) for 1 h and then incubated with the goat polyclonal anti-LC3A/B (abcam, Cambridge, UK) at 4 °C for 16 h. Membranes were probed with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. The immunoreactive proteins were detected using enhanced chemiluminescence reagent (SuperSignal West Pico chemiluminescence substrate; Pierce Biotechnology) and then exposed to X-ray film (FujiFilmCorp.).
Animal care
Twenty-eight Wistar male rats (weight of 180 g) were housed in plastic containers, kept at room temperature at 23 °C and artificial lighting of 24 h cycles consisting in 12 h light/12 h dark. Rats were fed with pellets (Laboratory Rodent Diet Harlan) and water ad libitum. All experiments were reviewed and approved by the Research and Experimental Animal Care Committees of the Instituto Nacional de Neurología y Neurocirugía de México.
In vivo treatment
Before intracranial implantation, RG2 cells were briefly trypsinized to detach them from culture bottles, centrifuged and resuspended in the corresponding medium. The cell suspensions were regularly shaken to avoid cell sedimentation and kept in a sterile vial at 4 °C until stereotactic implantation.
Orthotopic glioma model
For the rat RG2 glioma cell implantation model, Fischer 344 male rats (Bioinvert, Mexico City), 3-months-old (n = 28), were fed ad libitum. The cell implantation procedure was conducted based on the method developed by Kobayashi et al. (1980). Briefly, each animal was anesthetized (ketamine—40–90 mg/Kg intraperitoneally administered plus xylazine 5–10 mg/Kg subcutaneously administered) and immobilized on a stereotaxic unit (Stoelting Co., Wood Dale, IL). After disinfection and incision of the skin of the head with a scalpel, a hole was drilled through the skull 2 mm lateral and 2 mm anterior to the bregma, on the right-hand side of the skull. 1 × 106 rat glioma cells suspended in 10 μl of saline solution were injected at 3 mm in depth from the dura at a rate of 2 μl/min, using a 25-gauge Hamilton needle mounted on a 25-μl Hamilton glass syringe (Hamilton, Reno, NV). A waiting time of 2 min was implemented following injection.
After implant, animals were divided into four groups (n = 7 each), according to the treatment: the first group (control) was untreated, the second group (TMZ) was treated with temozolomide (10 mg/Kg in 200 μl of apple juice) administered by oral route for 5 days, the third group (PTx) was treated with 2 μg of PTx dissolved in 200 μl of saline solution 2 days after implant and 1 μg of PTx 4 days after implant, and the fourth group (TMZ + PTx) was treated with the combination of TMZ and PTx as previously mentioned.
Statistical analyses
For descriptive purpose, continuous variables were summarized as arithmetic means and standard deviations (SD). One-way analysis of variance and post hoc (Tukey) test were conducted. Statistical significance was determined with p < 0.05 in a two-sided test. Cumulative survival was determined from day of implantation until the rats showed disabling alterations, until death or the last day of follow-up, respectively. Data were analyzed by the Kaplan–Meier test. SPSS software package V 18.0 for Windows (SPSS Inc., Chicago, IL) was employed for data analysis.
Results
Cell viability and apoptosis induction in malignant RG2 glioma cell line
Figure 1a shows representative dot plots from cells treated with TMZ, PTx, and TMZ + PTx, respectively. The cytotoxicity effect of TMZ, PTx, or TMZ + PTx on RG2 glioma cell was determined by flow cytometric analysis assays to determine cell viability. After 48 h of exposure to treatments, a significant decrease in cell viability was observed in groups treated with TMZ (82.2 ± 0.91), PTx (83.1 ± 0.06), and TMZ + PTx (80.2 ± 0.65) compared to control group (96.2 ± 0.55) (p < 0.001). Figure 1b1.

TMZ or PTx effect on apoptosis induction in malignant RG2 glioma cells. Tumor cells were seeded at 3 × 105 cells per well (1 ml) in 6-well flat-bottomed plates and incubated overnight at 37 °C. After exposure to TMZ (100 mM), PTx (20 ng/ml) for 3 days, the cells were trypsinized, and the number of viable cells, necrotic, and apoptotic cells was determined by flow cytometry. Results showed the mean ± SD of three independent experiments. a Representative dot plots of apoptosis induction by TMZ, PTX, or TMZ + PTx. b Representative percentages of viability (b1), early apoptosis (b2), necrotic cells (b3), and late apoptosis (b4) from RG2 cells treated with TMZ, PTx, or TMZ + PTx
The degree of early and late apoptosis of glioma cells induced by TMZ, PTx, or TMZ + PTx was analyzed by FACS. The early apoptotic fraction in cells treated with TMZ (14.2 ± 1.05), PTx (12.9 ± 0.03), and TMZ + PTx (13.6 ± 0.95) was significantly higher than in control group (2.8 ± 0.15) (p < 0.001) after 48 h. Regarding late apoptosis induced in cells by the treatments, there were significant differences in TMZ (3.2 ± 0.21), PTx (3.2 ± 0.32), and TMZ + PTx (5.5 ± 1.11) groups compared to control group (0.81 ± 0.44) (p = 0.007 and p < 0.001). See Fig. 1b2, b3. We did not find differences between groups in the percentage of necrotic cells (p = 0.795). See Fig. 1b4. These results indicate that the observed cytotoxicity of TMZ or PTx is not due to necrosis induction.
Effect of TMZ, PTx, and TMZ + PTx treatments on cell cycle
To address more information whether the antiproliferative effects of TMZ, PTx, or their combination with malignant glioma are associated with cell cycle regulation, cell cycle analysis was carried out in RG2 cells treated with TMZ, PTx, or TMZ + PTx by DNA flow cytometric analysis. The TMZ treatment (100 mM, 48 h), PTx (20 ng/ml), and TMZ + PTx did not produce any influence in the induction of cell cycle in RG2 malignant glioma when treatments were administrated (data not shown).
Induction of autophagy in RG2 glioma cells
Is well know that radiation or chemotherapeutic agents such as tamoxifen, arsenic trioxide or TMZ induce autophagy, but not apoptosis in several cancer cells including malignant glioma cells (Kanzawa et al. 2004). To evaluate whether TMZ, PTx, or their combination induced morphological alterations compatible with autophagy, RG2 malignant glioma cells were exposed to TMZ (100 mM), PTx (20 ng/ml), or TMZ + PTx for 3 days. After that, chamber slides were stained with LC3 and Beclin, and the morphological changes were analyzed by microscopy. As shown in Fig. 2a, b, numerous autophagy vacuoles LC3 and Beclin 1 were observed in all the treatments. To determine autophagy activity, the expression of LC3 I and II (one of the autophagosome membrane proteins) was analyzed by Western blot (Fig. 2c). An increase in the amounts in both LC3-I and II was found in groups treated with TMZ, PTx, and TMZ + PTx. Similarly, up-regulated autophagy-related protein Beclin 1 was increased in groups that received these treatments (Fig. 2b).

Autophagy induction by TMZ, PTx, or TMZ + PTx in RG2 cells. Tumor cells were cultured on the chamber slide dish and incubated overnight at 37 °C. After exposure to TMZ (100 mM), PTx (20 ng/ml) for 3 days, cells were fixed and stained with the goat polyclonal anti-LC3A/B (a) or rabbit polyclonal anti-Beclin 1 antibody (b). Representative images of punctuate signal of a LC3 or b Beclin 1 in PTx, TMZ, or TMZ + PTx-treated RG2 cells were seen after treatment with TMZ (100 mM), PTx (20 ng/ml), or TMZ + PTx for 72 h. Cells were examined by fluorescence microscopy. c Western blot analysis of LC3 I and II expression in RG2 cells after 72 h of treatment with TMZ (100 mM), PTx (20 ng/ml), or TMZ + PTx
Development of acidic vesicular organelles (AVOs) in RG2 glioma cells
To detect and quantify the development of AVOs, we performed a flow cytometric analysis to determine whether TMZ, PTx, and TMZ + PTx could induce changes in fractional volume and/or acidity of AVOs. TMZ (7.0 ± 2.5), PTx (11.0 ± 6.5), and TMZ + PTx (49.0 ± 9.54) increased the strength of the bright red fluorescence (y-axis) in RG2 glioma cells from control (4.0 ± 1.30) after 48 h of treatment (Fig. 3b p < 0.05), respectively. These results could be attributable to the development of AVOs associated with autophagy.

Development of AVOs in TMZ, PTx, or TMZ + PTx-treated RG2 cells. Detection of green and red fluorescence in acridine orange-stained cells using FACS analysis (a). FL1-H indicates green color intensity, while FL3-H shows red color intensity. Graph shows the percentage of AVOs induction ± SD. Data shown are representative of three independent experiments. b For cell cycle analysis, malignant glioma cells treated with TMZ, PTx, or TMZ + PTx for 3 days were collected and stained with propidium iodide and analyzed in the FACSCalibur. The percentage of cells in different phases of the cell cycle was determined by using Flow jo Software version
Morphological changes and autophagy induction in RG2 cells
To reveal whether the observed cell damage was a cause or consequence of autophagy, the autophagy inhibitor, 3-methyladenine (3-MA, 1.0 mM), was administrated in RG2 cells treated with TMZ (100 mM), PTx (20 ng/ml), PTx (200 ng/ml), TMZ (100 mM) + PTx (20 ng/ml), or TMZ (100 mM) + PTx (200 ng/ml) after 24, 48, and 72 h of exposure.
After 24 h of exposure to treatments, some cells show vacuoles mainly in the groups treated with PTx (20 and 200 ng/ml) or in combination with TMZ (Fig. 4a). However, after 48 h of treatment, the number of cells showing autophagy membranes was increased, while the number of cells that maintain integrity in their membranes was reduced after addition 3-MA, an inhibitor of phosphatidylinositol-3 kinase (PI3K), which is known to inhibit autophagy sequestration (Fig. 4b). After 72 h of addition of 3MA, cells were stained with an anti-LC3 antibody, and we found that the treatment with TMZ, PTx, or TMZ + PTx diminished significantly the number of viable cells indicating an autophagy process stage (Fig. 4c).


Kinetics of autophagy induction. Kinetics of cell damage induced by TMZ, PTx (20, 200 ng/ml), TMZ + PTx (20 ng/ml), or TMZ + PTx (200 ng/ml) treatment after 24 h (a). The same treatments were tested with or without 3MA after 48 h (b) and 72 h (c). Images were taken in bright field and lather by fluorescence microscopy at 72 h posttreatment (d). Nuclei were counterstained with propidium iodide (red), and LC3 antibody was used to show cells expressing LC3 aggregation after treatment (green). 3MA was used to inhibit autophagy
TMZ + PTx increased survival in rats bearing intracerebral RG2 glioma
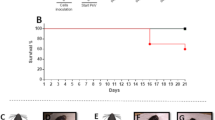
In rats bearing intracranial RG2 glioma tumors, the mean survival period determined was estimated from 19 to 21 days for the untreated group. A slight increase in the mean survival period was observed in the groups treated with TMZ (25 days p = 0.05) and PTx (28 days p = 0.048), but in the TMZ + PTx group, a synergistic effect was seen in the survival curve (31 days p = 0.014), suggesting that the combination of TMZ + PTx is effective for rat survival (Fig. 5).

Kaplan–Meier survival of rats with intracranial RG2 cells. Systemic (i.p.) administration of PTx (2 μg at day 0 and 1 μg at day 2), TMZ 10 mg/Kg/5 days (p.o), or TMZ + PTx prolonged survival of immunocompetent rats bearing intracranial RG2 glioma. Survival curves and Kaplan–Meier survival analysis of rats implanted with RG2 (1 × 106 cells/rat) and treated with TMZ, PTx, or TMZ + PTx (n = 7). The combination of TMZ + PTx potentiates rat survival
Discussion
Despite the multimodality treatments available, the prognosis of malignant glioma remains poor. Currently, TMZ is considered the most effective drug in the treatment for gliomas; however, its efficacy is often limited by tumor recurrence and the development of resistance to TMZ. Due to an unsatisfactory response of conventional treatment against gliomas, we tested a PTx treatment alone or in combination with the clinical alkylating agent, TMZ. Our results showed that the viability of RG2 cells was diminished in the groups treated with TMZ or PTx alone and also when these drugs were combined. However, when we analyzed the induction of apoptosis, we observed around 20 % of apoptotic cells in the groups treated with TMZ and PTx and we did not find any changes in the cell cycle arrest. These data agree with others previously reported by Sato et al. (2009) who did not observe significant changes in either apoptosis or cell cycle kinetics in treated glioma (T98G) cells. Similarly, Carmo et al. (2011) found that the expression of LC3, the autophagy-associated protein, was increased and only a reduced percentage of cells underwent apoptosis in TMZ-treated U-118 glioma cells. Conversely, Hirose et al. reported that in TMZ-treated glioma cells, the drug induced a cell cycle arrest in G2/M and a low level of apoptosis as compared with lymphoid cells. Other studies have proposed that the induction of G2/M arrest of malignant glioma cells occurs in a dose-dependent manner (Kanzawa et al. 2004; Hirose et al. 2001). We observed that TMZ decreased the level of apoptosis and also elicited the induction of the cell cycle by TMZ, which could be associated with the activity of O6-methylguanine-DNA methyltransferase (MGMT) and Pi3 K/Akt. It is known that the hypermethylation of the MGMT promoter reduces the ability to TMZ to induce apoptosis. Furthermore, an active state of Pi3 K/Akt overcomes the G2 checkpoint, thereby avoiding the effect of TMZ (Carmo et al. 2011; Law 2005; Meloche and Pouyssegur 2007; Tran et al. 2001). Similarly, we previously reported that PTx treatment is capable to decrease the cell viability and induce around 25 % of apoptosis in the glioma C6 model (Orozco-Morales et al. 2012).
Regarding to the low level of induction of apoptosis, we determined whether the observed autophagy was induced in RG2 cell after treatment with TMZ and PTx; therefore, we analyzed the level of expression of LC3 I, II, Beclin 1, and the formation and promotion of AVOs after 3MA addition. We found that TMZ and PTx treatments elicited autophagic cell death characterized by the high level of autophagic protein Beclin-1 and a cellular redistribution of the marker LC3. Notoriously, this effect was increased when TMZ + PTx was combined in the treatment. Furthermore, after 3MA was added, we found a decrease in the number of autophagic vesicles and the formation of autophagosome, in the groups treated with TMZ, PTx, and TMZ + PTx. The mechanisms of action of TMZ and the pathways involved in the escape of glioma cells from death have not yet been completely elucidated. However, it has been proposed that TMZ is able to induce autophagy which can be either survival promoting or death inducing depending on the cellular context (Kanzawa et al. 2003b; Thorburn 2008). In the context of brain tumors, cytoplasmic levels of the protein BECLIN1 and its mRNA were found to be lower in GBMs compared to lower-grade astrocytomas as well as in normal brain tissue (Miracco et al. 2007). Additionally, high cytoplasmic levels of the protein BECLIN1 have been found to show a positive correlation with the survival and the performance status of patients, whereas a low expression of BECLIN1 correlates with an increase in cell proliferation and a decrease in apoptosis (Huang et al. 2010). Interestingly, high levels of expression of LC3 have been associated with improved survival in GBM patients with poor performance scores, whereas in patients with normal performance scores, low levels of expression of LC3 correlate with better survival rates (Aoki et al. 2008). It has been described that low levels of BECLIN1 and LC3B-II proteins have been found in higher grade astrocytomas (Huang et al. 2010), suggesting that a decrease in autophagic activity may drive the progression of astrocytic tumors.
Subsequently, we perform an in vivo study in order to determine whether the treatments for TMZ, PTx, or TMZ + PTx could increase the survival of rats implanted intracranially with RG2 glioma cells (Fig. 5). Our results demonstrated a significant increase in survival when animals were treated both with TMZ and PTx. However, the combined treatment with TMZ + PTx increased two times more the survival registered compared when the treatments were administered individually. Our group previously demonstrated that the treatment with PTx alone reduces the subcutaneous glioma size until 80 % compared to the control group. Furthermore, we demonstrated that PTx was capable to induce selectively apoptosis of Treg cells, and it elicited a decrease in the number of tumor-invading macrophages (Orozco-Morales et al. 2012). It is possible that the dual effect observed for PTx over the modulation of immune response and autophagy induction produces an increase in the survival of rats intracranially implanted with RG2 glioma cells. We attribute this effect to the immunomodulation produced by PTx plus the cytotoxic effect and autophagy induction by TMZ. Additionally, PTx exerts pleiotropic effects on the immune system, and it plays an important role in the activation of toll-like receptor 4 (TLR4). Recently, it was shown that TLR 4 acts as an environmental sensor for autophagy, and recent data have revealed that the regulation of TLR4 in several glioma cell lines plays an important role in the autophagy response, tumor progression, and chemoresistance (Sarrazy et al. 2011; Yu et al. 2012; Tewari et al. 2012; Waltz et al. 2011; Xu et al. 2007; Magaña et al. 2012). It is possible that PTx caused irreversibly autophagy activating the cell death by TLR4, increasing the efficacy given by TMZ, which may be a pivotal mechanism in mediating Pi3 K/Akt and ERK1/2 MAP kinase signaling pathways (Ogier-Denis and Codogno 2003; Marino and Lopez-Otin 2004; Fu et al. 2010; Thorburn 2008; Katayama et al. 2007). Furthermore, PTx is frequently used in immunological studies to enhance the onset of autoimmune disease in experimental animals (Locht et al. 2011). PTx-induced permeability of cerebral endothelial barriers is mediated through the PKC and PI3-kinase pathways, and it is abrogated by high cAMP levels (Bruckener et al. 2003). Therefore, it is possible that the multiple effects of the PTx over the immune system, autophagy induction and blood brain barrier permeabilization with consequent increase in the amount of TMZ inside the brain tumor, could warrant the use of PTx in combination with the actual regimen of chemotherapy.
In conclusion, the concomitant treatment with TMZ and PTx induced autophagy in vitro and increased the survival in RG2 glioma model studied here.
References
Aoki H, Kondo Y, Aldape K, Yamamoto A, Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K, Shinojima N, Shingu T, Tamada Y, Zhang L, Conrad C, Bogler O, Mills G, Sawaya R, Kondo S (2008) Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 4(4):467–475
Bruckener KE, el Baya A, Galla HJ, Schmidt MA (2003) Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J Cell Sci 116(Pt 9):1837–1846
Buckner JC (2003) Factors influencing survival in high-grade gliomas. Semin Oncol 30(6 Suppl 19):10–14
Carmo A, Carvalheiro H, Crespo I, Nunes I, Lopes MC (2011) Effect of temozolomide on the U-118 glioma cell line. Oncol Lett 2(6):1165–1170. doi:10.3892/ol.2011.406ol-02-06-1165
Carracedo J, Ramirez R, Marchetti P, Pintado OC, Baixeras E, Martinez C, Kroemer G (1995) Pertussis toxin-sensitive GTP-binding proteins regulate activation-induced apoptotic cell death of human natural killer cells. Eur J Immunol 25(11):3094–3099. doi:10.1002/eji.1830251116
Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA (1994) NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 33(31):9045–9051
Deorah S, Lynch CF, Sibenaller ZA, Ryken TC (2006) Trends in brain cancer incidence and survival in the United States: surveillance, Epidemiology, and End Results Program, 1973 to 2001. Neurosurg Focus 20(4):E1. doi:10.3171/foc.2006.20.4.E1
Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP (2010) Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol 12(4):328–340. doi:10.1093/neuonc/nop005
Hirose Y, Berger MS, Pieper RO (2001) p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res 61(5):1957–1963
Huang X, Bai HM, Chen L, Li B, Lu YC (2010) Reduced expression of LC3B-II and Beclin 1 in glioblastoma multiforme indicates a down-regulated autophagic capacity that relates to the progression of astrocytic tumors. J Clin Neurosci 17(12):1515–1519. doi:10.1016/j.jocn.2010.03.051
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19(21):5720–5728. doi:10.1093/emboj/19.21.5720
Kanzawa T, Bedwell J, Kondo Y, Kondo S, Germano IM (2003a) Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg 99(6):1047–1052. doi:10.3171/jns.2003.99.6.1047
Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I (2003b) Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63(9):2103–2108
Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S (2004) Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 11(4):448–457. doi:10.1038/sj.cdd.44013594401359
Katayama M, Kawaguchi T, Berger MS, Pieper RO (2007) DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 14(3):548–558. doi:10.1038/sj.cdd.4402030
Kim J, Klionsky DJ (2000) Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem 69:303–342. doi:10.1146/annurev.biochem.69.1.303
Kobayashi N, Allen N, Clendenon NR, Ko LW (1980) An improved rat brain-tumor model. J Neurosurg 53(6):808–815. doi:10.3171/jns.1980.53.6.0808
Law BK (2005) Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol 56(1):47–60. doi:10.1016/j.critrevonc.2004.09.009
Locht C, Coutte L, Mielcarek N (2011) The ins and outs of pertussis toxin. FEBS J 278(23):4668–4682. doi:10.1111/j.1742-4658.2011.08237.x
Lopez-Gonzalez MA, Sotelo J (2000) Brain tumors in Mexico: characteristics and prognosis of glioblastoma. Surg Neurol 53(2):157–162
Magaña R, Hernandez-Pedro N, Rangel E, Manoutcharian K, Pineda B, Sotelo J (2012) Pertussis: symptoms, prevention and treatment. Adv Med Biol 55:41–68
Marino G, Lopez-Otin C (2004) Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci 61(12):1439–1454. doi:10.1007/s00018-004-4012-4
Meloche S, Pouyssegur J (2007) The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26(22):3227–3239. doi:10.1038/sj.onc.1210414
Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, Falzarano SM, Pirtoli L, Tosi P (2007) Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol 30(2):429–436
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152(4):657–668
Munafo DB, Colombo MI (2001) A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 114(Pt 20):3619–3629
National Cancer Institute (2011) Central brain tumor registry of the United States
Ogier-Denis E, Codogno P (2003) Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 1603(2):113–128
Orozco-Morales M, Sanchez-Garcia FJ, Guevara-Salazar P, Arrieta O, Hernandez-Pedro NY, Sanchez-Garcia A, Perez-Madrigal R, Rangel-Lopez E, Pineda B, Sotelo J (2012) Adjuvant immunotherapy of C6 glioma in rats with pertussis toxin. J Cancer Res Clin Oncol 138(1):23–33. doi:10.1007/s00432-011-1069-y
Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61(2):439–444
Pietra G, Mortarini R, Parmiani G, Anichini A (2001) Phases of apoptosis of melanoma cells, but not of normal melanocytes, differently affect maturation of myeloid dendritic cells. Cancer Res 61(22):8218–8226
Ryu CH, Yoon WS, Park KY, Kim SM, Lim JY, Woo JS, Jeong CH, Hou Y, Jeun SS (2012) Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J Biomed Biotechnol 2012:987495. doi:10.1155/2012/987495
Sarrazy V, Vedrenne N, Billet F, Bordeau N, Lepreux S, Vital A, Jauberteau MO, Desmouliere A (2011) TLR4 signal transduction pathways neutralize the effect of Fas signals on glioblastoma cell proliferation and migration. Cancer Lett 311(2):195–202. doi:10.1016/j.canlet.2011.07.018
Sato Y, Kurose A, Ogawa A, Ogasawara K, Traganos F, Darzynkiewicz Z, Sawai T (2009) Diversity of DNA damage response of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol Ther 8(5):452–457
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352(10):987–996. doi:10.1056/NEJMoa043330
Stupp R, Hegi ME, Gilbert MR, Chakravarti A (2007) Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol 25(26):4127–4136. doi:10.1200/JCO.2007.11.8554
Tewari R, Choudhury SR, Ghosh S, Mehta VS, Sen E (2012) Involvement of TNFalpha-induced TLR4-NF-kappaB and TLR4-HIF-1alpha feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med (Berl) 90(1):67–80. doi:10.1007/s00109-011-0807-6
Thorburn A (2008) Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 13(1):1–9. doi:10.1007/s10495-007-0154-9
Tran SE, Holmstrom TH, Ahonen M, Kahari VM, Eriksson JE (2001) MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Biol Chem 276(19):16484–16490. doi:10.1074/jbc.M010384200M010384200
Velasquez-Perez L, Jimenez-Marcial ME (2003) Clinical-histopathologic concordance of tumors of the nervous system at the Manuel Velasco Suarez National Institute of Neurology and Neurosurgery in Mexico City. Arch Pathol Lab Med 127(2):187–192
Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, Zuckerbraun BS (2011) Lipopolysaccharide induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 7(3):315–320. doi:10.4161/auto.7.3.14044
Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65(22):3640–3652. doi:10.1007/s00018-008-8383-9
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27(1):135–144. doi:10.1016/j.immuni.2007.05.022
Yu L, Wang L, Chen S (2012) Exogenous or endogenous Toll-like receptor ligands: which is the MVP in tumorigenesis? Cell Mol Life Sci 69(6):935–949. doi:10.1007/s00018-011-0864-6
Acknowledgments
Animals were kindly donated by Bioinvert Mexico City. This work was supported by the National Council of Science and Technology of Mexico (CONACyT, Grant 180851) and Instituto de Ciencia y Tecnología del Distrito Federal (PICSA 10-143).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Magaña-Maldonado, R., Manoutcharian, K., Hernández-Pedro, N.Y. et al. Concomitant treatment with pertussis toxin plus temozolomide increases the survival of rats bearing intracerebral RG2 glioma. J Cancer Res Clin Oncol 140, 291–301 (2014). https://doi.org/10.1007/s00432-013-1565-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-013-1565-3