
Abstract
Toll-like receptors (TLRs) are a class of pattern recognition receptors sensing microbial components and triggering an immune response against pathogens. In addition to their role in anti-infection immunity, increasing evidence indicates that engagement of TLRs can promote cancer cell survival and proliferation, induce tumor immune evasion, and enhance tumor metastasis and chemoresistance. Recent studies have demonstrated that endogenous molecules or damage-associated molecular patterns released from damaged/necrotic tissues are capable of activating TLRs and that the endogenous ligands-mediated TLR signaling is implicated in the tumor development and affects the therapeutic efficacy of tumors. Since both exogenous and endogenous TLR ligands can initiate TLR signaling, which is the most valuable player in tumor development becomes an interesting question. Here, we summarize the effect of TLR signaling on the development and progression of tumors, and discuss the role of exogenous and endogenous TLR ligands in the tumorigenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition proteins that detect both microbe- and host-derived molecular patterns. TLRs are expressed mainly on macrophages, dendritic cells (DCs) and epithelial cells. Ligand binding to TLRs activates transcription factors, leading to production of inflammatory mediators. Thus, TLRs play a crucial role in the innate immune response and the subsequent induction of adaptive immune response against pathogen infection. In recent years, increasing evidence demonstrates that functional TLRs are also expressed on tumor cells and the engagement of these TLRs induces production of proinflammatory cytokines and plays an active role in carcinogenesis and tumor progression during chronic inflammation. Moreover, accumulating evidence points out that the endogenous stimulators can activate TLR signaling and contribute to the pathogenesis of the tumor. Here, we review the role of TLR signaling as well as exogenous and endogenous TLR ligands in tumorigenesis.
Toll-like receptor signaling and damage-associated molecular patterns
So far, at least 11 mammalian TLRs have been identified, each of them recognizing distinct pathogen-associated molecular patterns (PAMPs) derived from various microorganisms (Table 1). TLRs 1, 2, 4, 5, and 6 are expressed and bind their ligands on the cell surface. TLRs 3, 7, 8, and 9 are nucleic acid receptors expressing on endosomal membrane. The ligand for TLR10 remains unknown [1]. Mouse TLR11 recognizes profilin-like molecule from the protozoan parasite [2], whereas Human TLR11 fails because of the presence of a stop codon in the gene [3]. Extracellular components MD-2 [4] and CD14 [5] are also critical for the recruitment of TLR4 ligands.
Most TLRs share a common signaling pathway in which myeloid differentiation factor 88 (MyD88) and toll/interleukin-1 receptor (TIR) domain-containing protein (TIRAP)/MyD88 adaptor-like (Mal) are recruited to activate tumor necrosis factor receptor-associated factor-6 (TRAF6)/IκB kinase (IKK) complex and mitogen-activated protein kinase (MAPK) kinase, respectively, resulting in early activation of the transcription factor NF-κB and MAPK (see reviews in [6, 7]). This is called MyD88-dependent TLR signaling pathway. Another two TIR-containing adaptor molecules, TIR domain-containing adaptor inducing interferon-β (TRIF)/TIR domain-containing adaptor molecule-1 (TICAM-1) and TRIF-related adaptor molecule (TRAM)/TIR domain-containing adaptor molecule-2 (TICAM-2), mediate the MyD88-independent signaling pathway leading to the activation of the late phase NF-κB and IFN regulatory factor 3 (IRF3) and the subsequent production of type I IFN (IFN α/β), IFN-inducible gene products, and an immune regulatory response [7].
In addition to the exogenous ligands of microbes, increasing evidence indicates that TLRs are receptors for the endogenous stimulators released from damaged tissues and mediate a non-infectious inflammatory response [8, 9]. These host-derived endogenous TLR ligands are either components of cells or induced gene products in specific conditions, including extracellular matrix components and intracellular proteins and nucleic acids (Table 1). Usually, endogenous TLR ligands do not interact physiologically with corresponding receptors because of their different cellular localization. In some pathological conditions, these endogenous molecules are released from injured/inflamed tissues and cells or actively secreted by inflammatory cells via a non-conventional lysosomal route [10, 11]. They activate TLRs and initiate a protective inflammatory response and the repair of damaged tissues. Thus, endogenous TLR ligands are often referred to as alarmins and serve as early warning signals to host immune systems. As these host-derived non-microbial molecules are released following tissue injury and cell death and are able to elicit similar responses as PAMPs, they have been collectively categorized as damage-associated molecular patterns (DAMPs). The activation of the immune system is therefore not only based on the recognition of PAMPs but also relies on the presence of DAMPs released by injured cells [12]. The endogenous TLR ligands-mediated sterile inflammation has been linked to a variety of pathological processes, especially tumorigenesis [13, 14].
Toll-like receptor signaling and tumors
Increasing evidence supports that TLR signaling plays an important role in tumorigenesis. At first, polymorphisms in TLR genes are related to risk of several cancers [15–17]. Some variations in TLR genes are even functionally associated with cancer susceptibility [18–20]. Importantly, TLR signaling-mediated chronic inflammation is involved in tumor development and contributes to tumor progression and chemoresistance [21, 22]. Moreover, the activation of TLR signaling could regulate antitumor immunity of the host [23] or induce tumor immune evasion [21, 24].
Polymorphism in Toll-like receptor genes and risk of human cancer
TLR2 gene polymorphisms have been associated with the risk of several human malignancies such as gastric cancer [25], colorectal cancer [15], cervical cancer [16], gallbladder cancer [26] and leukaemia [27, 28]. For example, TLR 2 (−196 to −174 del) gene polymorphism increases the risk of gastric cancer in the Japanese population [25] and increases the susceptibility of cervical cancer and gallbladder cancer in the Indian population [16, 26]. This allele also affects viral loads and increases the risk for hepatocellular carcinoma in patients with chronic hepatitis C [18]. It is believed that the deletion disrupts TLR2 promoter activity and the production of cytokines [18].
Asp299Gly polymorphism of the TLR4 gene is associated with the risk of sporadic colorectal cancer [15], mucosa-associated lymphoid tissue lymphoma and Hodgkin’s lymphoma [28]. The TLR4 Thr399Ile polymorphism is significantly associated with gallbladder cancer [26] and cervical cancer susceptibility in the Indian population [16]. Both TLR4 Asp299Gly and Thr399Ile polymorphisms are related to increased risk of precancerous gastric lesions and, possibly, gastric cancer [29–31]. The TLR4+896 A/G polymorphism impairs TLR4 reactivity to bacterial LPS and is a risk factor for noncardia gastric carcinoma [19]. A heterozygous Thr135Ala polymorphism at LRR motif of TLR4 has been reported in patients with poorly-differentiated gastric adenocarcinomas. The mutation of Thr to Ala may affect phosphorylation of TLR4 protein, providing a specific example that a functional polymorphism of TLR4 gene increases cancer risk [20]. In addition, TLR4+3725 G/C polymorphism is a risk factor of severe gastric atrophy in H. pylori-seropositive Japanese [32]. The several single-nucleotide polymorphisms (SNPs) in TLR4 gene increase the risk of prostate cancer in different populations [33–36]. TLR4 coreceptor CD14 promoter-159 polymorphism is associated with reduced risk of intestinal-type gastric cancer in a Japanese population [37].
It has been reported that the combination of two TLR9 variant alleles is protective for endometrial cancer risk [38]. TLR9-1237C and 2848A SNPs and TLR9/1237C-2848A haplotype are associated with an increased risk for Hodgkin’s lymphoma [39]. TLR3 polymorphism is an independent prognostic marker for stage II colorectal cancer [40], and the polymorphisms of both TLR3 and TLR10 are associated with nasopharyngeal carcinoma risk in the Cantonese population [17, 41]. Variation in the TLR10-TLR1-TLR6 gene cluster is related to the risk of prostate cancer [42] and non-Hodgkin lymphoma [27].
Gene polymorphism may only represent a disease-associated marker, but the functional association of TLR polymorphisms with cancer risk illustrates an important role of TLRs in tumorigenesis.
Toll-like receptor signaling-mediated tumorigenesis and antitumor immunity
A number of studies report that functional TLRs are widely expressed on cancers or cancer cell lines, suggesting a critical role of TLRs in tumors [21, 24, 43, 44]. Knockdown of TLR4 gene results in a dramatic reduction of human breast cancer cell viability [45] and efficiently inhibits established tumor growth in vivo in a mouse prostate cancer model [46]. Deficiency of TLR4 signaling in mice enhances cancer-related survival and reduces tumor growth in the lungs [14], and protects mice from colon carcinogenesis [47]. Tumor cells also utilize the TLR4 signaling pathway to escape immune surveillance and enhance tumor cell invasion and metastasis. For example, activation of TLR4 in tumor cells results in resistance of tumor cells to CTL attack [21]. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance [24]. Triggering of TLR4 expressed on human head and neck squamous cell carcinoma (HNSCC) protects the tumor from immune attack and supports tumor progression [43]. Knockdown of TLR4 in human prostate cancer cells results in a dramatic reduction of tumor cell migration and invasion [46]. TLR4 signaling also promotes immune escape, progression and chemoresistance of ovarian cancer [22, 44]. Thus, activation of TLR4 on tumor cells appears to promote tumor cell proliferation, resistance to apoptosis and immune escape.
The pro-tumorigenic effects of TLR4 signaling are involved in activation of NF-κB and many other inflammatory mediators in tumor cells and immune cells. Inappropriate TLR4 signaling results in chronic inflammation, which leads to the increase of NF-κB activity in cancer cells to drive cancer growth. Activation of NF-κB in both tumor cells and tumor-associated macrophages (TAMs) is dependent on TLR4 signaling [14]. TNF-α produced by macrophages mediates NF-κB activation in tumor cells, and in turn tumor cells could induce NF-κB activation in macrophages [14]. The activation of TLR4 on human colon cancer cells can increase the phosphorylation of ERK, regulate the expression of IL-8 and caspase-7, and promote the proliferation and migration of human colon cancer cells [48]. In addition, TLR4 signaling has been found to be responsible for induction of cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2) production, and EGFR phosphorylation, which promote the development of colitis-associated colorectal tumors [47, 49–52]. Expression of mucosal PGE2 is decreased in TLR4-deficient mice and TLR4-deficiency protects mice from colitis-associated neoplasia. Exogenous administration of PGE2 in TLR4-deficient mice increases colitis-associated tumor incidence and tumor size. These data suggest that PGE2 is a central downstream molecule involving TLR4-mediated intestinal tumorigenesis [53].
In contrast to TLR4, TLR2 signaling protects mice from tumor development of colitis-associated colorectal cancer (CAC) [54]. TLR2-deficient CAC mice develop significantly more and larger colorectal tumors than CAC littermates with wild-type (WT) TLR2. TLR2-deficient colons have more advanced dysplasia compared to WT colons. TLR2-deficiency leads to increased early formation of aberrant crypt foci and early intestinal tumorigenesis. TLR2-deficiency increases cell proliferation and reduces apoptosis during early CAC development. TLR2-deficient mice show increased IL-6 and STAT3 activation during early intestinal tumorigenesis and have an increased Th17 immune response during CAC development. Colonic tissues from TLR2-deficient mice recruit inflammatory cells that have reduced nitric oxide (NO) production and fail to mount an adequate defense against tumor growth, which leads to inflammatory growth signals and predisposition to accelerated neoplastic growth [54]. Interestingly, more immune cells express TLR2 in oral squamous cell carcinoma and dysplasia than in hyperplasia. No hyperplastic samples show positive TLR2 staining on keratinocytes, whereas keratinocytes in 64% of cases of carcinoma and 74% of cases of dysplasia are TLR2 positive [55]. Although the authors suppose that TLR2 expression in the tumor microenvironment may indicate activation of immune surveillance against the altered epithelium, whereas TLR2 expression by malignant keratinocytes is probably indicative of resistance to apoptosis as a pro-survival mechanism, the result seems inconsistent with what has been observed in colorectal cancer.
Activation of TLR3 on human cancer cells with poly(I:C) inhibits cell proliferation, triggers apoptosis [56–58], and keeps tumors more differentiated [59]. Implanted prostate tumors in TLR3-defecient mice grow markedly larger compared with WT mice, and type I Interferons contribute to TLR3-mediated tumor suppression [59]. Treatment with polyadenylic–polyuridylic acid [poly(A:U)], which only signals through TLR3, induces the secretion of a large amounts of CCL5/RANTES and CXCL10/IP-10 by mouse melanoma and glioma cells, primary human breast cancer cells, and tumor in a mouse model, suggesting that poly(A:U) can directly trigger TLR3 on tumor cells to stimulate the production of chemokines both in vitro and in vivo [60].
It has been reported that functional expression of TLR9 promotes cell proliferation and survival in human hepatocellular carcinomas [61]. Synthetic TLR9 ligands induce tumor invasion in human breast, prostate and lung cancer cells in vitro [62–66]. TLR 9 agonist CpG-ODNs up-regulates the expression of COX-2 and promotes the expression and secretion of immunosuppressive cytokines TGF-β1 and IL-8 through NF-κB activation in human prostate cancer cells, which may be implicated in tumor invasion and metastasis [67, 68]. However, CpG-ODNs stimulation inhibits the proliferation of TLR9-expressing neuroblastoma (NB) cells, induces caspase-dependent apoptotic cell death, and significantly prolongs the survival of mice bearing NB tumor xenografts [69]. TLR9 expression in primary human NB specimens has been found to correlate inversely with disease stage [69]. TLR9 expression in bone marrow cells of myelodysplastic syndromes is also down-regulated during transformation to overt leukemia [70]. The role of TLR9 signaling in tumorigenesis appears dependent on the tumor type.
Upon the activation most TLRs recruit adaptor MyD88 for the downstream signal. Oncogenic MyD88 mutations have been associated with the activated B-cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) [71]. Substitution of Leu265Pro at an evolutionarily conserved residue in the hydrophobic core of MyD88 TIR domain is observed in 29% of ABC DLBCL, whereas the mutation is rare or absent in other DLBCL subtypes and Burkitt’s lymphoma. The Leu265Pro substitution promotes cell survival by spontaneously assembling a protein complex containing IRAK1 and IRAK4, leading to activation of downstream signaling of MyD88 and secretion of cytokines [71].
It seems that each TLR signaling has its particular effect on tumors. Outcomes of TLR activation not only rely on receptors but also vary according to different tumor types. As we described above, activation of TLR4 shows a pro-tumorigenic effect in most tumor types such as prostate and lung cancers [14, 46]. But mice with a mutated TLR4 develop more skin tumors than WT mice treated with a mutagenic agent, suggesting that TLR4-dependent antitumor responses are important for inhibiting tumorigenesis in skin [72]. However, the underlying mechanism has not been well investigated.
In addition to triggering of TLRs in tumor cells, TLR-stimulated immune cells play a pivotal role in tumor development and progression. For example, transfer of macrophages with WT TLR4 to TLR4-deficient mice with experimental metastatic melanoma reduces survival time, but it has no effect on WT TLR4 recipient mouse, suggesting that functional TLR4 on macrophages can enhance tumor growth [14]. TLR3 engagement promotes DC maturation and T cell activation [73]. Treatment of human DC in vitro with poly(I:C12U) (rintatolimod), a modified form of poly(I:C), results in up-regulation of a number of DC markers, such as CD86 and MHC molecules, which are central to antigen presentation and immune cell activation [73]. Poly(I:C12U) induces the release of various proinflammatory chemokines and cytokines by DC, which are associated with specific T cell responses and immune regulation. Poly(I:C12U) boosts the DC mediated antigen-specific T cell response in vitro. Importantly, poly(I:C12U) significantly enhances prostate specific antigen-specific T cell and antibody responses in a prostate-specific antigen transgenic mouse model [73]. TLR3 activation of peripheral blood mononuclear cells significantly enhances antibody-dependent cellular toxicity against tumors [73]. In addition, when poly(I:C)- or imiquimod-treated tumor cells are cocultured with γδ T cells, activation of TLR3 or TLR7 on tumor cells enhances cytotoxic activity of γδ T cells of cancer patients and tumor cell lysis by human γδ T cells [74]. Moreover, in a mouse xenograft model of human colon cancer, it has been revealed that the production of neutrophil attracting chemokines and ensuing neutrophil infiltration are dramatically reduced in TLR5-deficient tumor xenografts, indicating that TLR5 signaling could be a potential immunotherapeutic target [75]. The roles mediated by different TLR are summarized in Table 2.
Due to the importance of TLR signaling in tumorigenesis, TLR agonists have potential for antitumor therapy. Poly(A:U) has been used for treating breast or gastric cancers [76, 77]. Imiquimod, a synthetic TLR7 ligand, was licensed in 1997 by the US Food and Drug Administration for the treatment of malignant tumors of the skin and genital warts caused by human papilloma virus (HPV) infection [78]. TLR9 ligand, CpG-ODNs, has been used in a phase I trial of non-Hodgkin’s lymphoma [79]. Several other synthetic TLR ligands are in clinical trial for treatment of different cancers.
Expression of Toll-like receptors is associated with the progression and prognosis of cancer
Since TLR signaling is involved in tumorigenesis, the expression of TLRs on tumor cells is associated with progression of cancer and disease prognosis. Consistent with pro-tumorigenic effects of TLR4, the high expression of TLR4/MyD88 in colorectal cancer (CRC) is associated with liver metastasis, earlier relapse, and poor disease-free survival and overall survival. Thus, TLR4 is an independent predictor of poor prognosis in patients with CRC [51, 80]. However, TLR4 expression gradually diminishes as the prostate cancer becomes more aggressive [81]. We have observed a decreased expression of TLR4 during the progression of cervical neoplasia and have found that the down-regulation of TLR4 appears to be associated with the expression of P16INK4A which is a crucial marker of HPV integration into host cells [82]. It is most likely that infection factors such as HPV are involved in the regulation of TLR4 expression.
The expression of TLR9 is associated with poor differentiation in prostate [83], breast, and ovarian cancers [84]. It has been found that TLR9 overexpression and stimulation with hypomethylated DNA augment the migratory capacity of cancer cells [84]. In addition, highly metastatic human breast cancer cells express high levels of TLR2 in contrast to poorly metastatic breast cancer cells and homogenous untransformed breast cells [85]. TLR8 expression is also an independent prognostic factor for CRC [86]. Prostate carcinomas with high TLR3 or TLR9 expression levels exhibit a higher probability of biochemical recurrence [87]. Moreover, the expression levels of TLR3, TLR4 and TLR9 may indicate the aggressiveness of human breast cancer. For example, the tumors with high TLR3 expression by tumor cells or with high TLR4 expression by mononuclear inflammatory cells are significantly associated with a higher probability of metastasis [88].
The role of exogenous and endogenous Toll-like receptor ligands in tumorigenesis
TLR signaling plays a critical role in tumorigenesis, but the key question is what has triggered TLR signaling. According to the current evidence, it appears that both exogenous and endogenous TLR stimulators contribute to the activation of TLRs.
Exogenous Toll-like receptor ligands
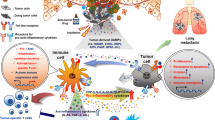
It is well known that chronic bacterial infection can increase the risk of many solid tumors. Usually, this is attributed to bacterial-caused inflammation, but the underlying mechanism has not been well evaluated. Recent studies demonstrate that microbial components, serving as exogenous TLR ligands to trigger TLR signaling, induce the production of cytokines and proangiogenic factors, promote invasiveness and adhesiveness of the cancer cells, and enhance tumor metastasis, which play an important role in chronic inflammation-promoting tumor development and progression (Fig. 1).

Potential effects of different TLR ligands on tumor progression and host immunity
TLR4 ligand LPS is one of key mediators. TLR4 activation in tumor cells by LPS induces the synthesis of various protein factors or proinflammatory cytokines, promotes tumor growth, and results in resistance of tumor cells to CTL attack and paclitaxel [21, 22]. LPS stimulation activates the NF-κB in many tumor types, induces the production of immunosuppressive and proangiogenic cytokines such as TGF-β, VEGF and IL-8, enhances apoptosis resistance, and promotes immune escape and metastasis [24, 44, 89]. These findings clearly link LPS/TLR4 signaling to inflammation, tumor growth, immune escape, and chemoresistance. It has been reported that phosphatidylinositol-3′-kinase (PI3K)/Akt signaling pathway is involved in CRC growth and progression [90]. TLR4 activation treated with LPS increases Akt phosphorylation in colon cancer cells [91, 92]. LPS stimulation increases adherence of CRC cells in vitro and in vivo, and enhances liver metastasis of human TLR4-expressing CRC cells after intrasplenic graft of nude mice [92]. Enhanced adherence induced by LPS could be blocked by TLR4 antagonist, PI3K inhibitor, or anti-β1 integrin blocking antibodies. The results indicate that LPS-induced TLR4 signaling activates the PI3K/Akt pathway and promotes downstream β1 integrin function, thereby increasing the adhesiveness and metastatic capacity of CRC cells. This may be the reason that infectious complications resulting from resection of CRC elevate the risk of cancer recurrence and metastasis [92].
The fact that gut-derived LPS amplifies the tumorigenic response of the liver further supports a role of exogenous TLR ligand in tumor development [93]. The circulating levels of LPS are elevated during tumor progression in animal models of diethylnitrosamine (DEN)-induced hepatocarcinogenesis, indicating that plasma endotoxin may be a critical cofactor in chemically induced hepatocarcinogenesis [93]. Depletion of host microflora in rats treated with antibiotics significantly reduces the levels of LPS in animal plasma, the production of TNF-α and IL-6, and liver fibrogenesis after DEN treatment. The number of detectable tumors, maximal diameters of tumors and the relative liver weight significantly decrease in rats treated with antibiotics and DEN. Ki-67 immunostaining indicates that antibiotic-treated rats have a significantly lower level of cell proliferation. Similarly, genetic deficiency of TLR4 in mice greatly decreases chemically induced hepatocarcinogenesis. These data suggest that the microbial LPS contributes to tumor induction after DEN treatment and that sustained LPS accumulation represents a pathological mediator of inflammation-associated hepatocellular carcinoma [93]. The pro-tumorigenic effect of endotoxin is mainly due to elevated NF-κB activity in premalignant epithelial cells, which suppresses apoptosis, thus promoting the cell survival and subsequent tumor development [93].
It has been found that Listeria monocytogenes (Lm) infection promotes tumor growth in a TLR2-dependent manner. Lm activates MAPKs and NF-κB in tumor cells through TLR2 signaling, resulting in the increased production of NO and IL-6 and increased proliferation of tumor cells [94]. Activation of TLR2 in breast cancer cells by peptidoglycan (PGN) from infectious bacterium Staphylococcus aureus (PGN-SA) induces TLR2-dependent NF-κB activation and secretion of IL-6 and TGF-β and promotes the invasiveness and adhesiveness of the cancer cells in vitro, illustrating a new link between component of infectious bacteria and the cancer [85]. PGN-SA-stimulated NF-κB activity results in STAT3 and Smad3 sequential activation, which contributes to invasiveness and adhesiveness [85]. STAT3 and Smad3 were identified as important factors responsible for malignant tumor progression [95, 96].
The role of flagellin-mediated TLR5 signaling in tumor development is controversial. Flagellin enhances the proliferation of human gastric cancer cells in a TLR5-dependent manner [97]. But flagellin-activated TLR5 signaling in breast cancer cells inhibits cell proliferation and an anchorage-independent growth, exhibiting a potent antitumor activity [98]. The in vivo inhibitory effect has been confirmed in mouse xenografts of human breast cancer cells [98]. In addition, knockdown of TLR5 expression dramatically enhances tumor growth and inhibits tumor necrosis in mouse xenografts of human colon cancer. Activation of TLR5 by flagellin substantially increases tumor necrosis, leading to significant tumor regression [75]. A further study on other tumor types will be valuable for the evaluation of flagellin/TLR5 signaling.
Endogenous Toll-like receptor ligands
In addition to exogenous ligands from microorganisms, TLRs recognize the endogenous molecules released from injured tissues, triggering an inflammatory response to increase the risk of cancer. Thus, understanding endogenous ligands-mediated TLR signaling in tumorigenesis is becoming an important issue for tumor prevention and therapy. It has been reported that dying tumor cells release endogenous TLR ligands that are involved in tumor progression [14, 23].
HMGB1 is a critical mediator of ischemia-induced inflammation. In the case of tumors, HMGB1 recognition has a paradoxical dual effect: promoting tumor development or triggering anti-neoplastic immune responses [13, 23]. HMGB1 released by necrotic keratinocytes recruits inflammatory cells and triggers a TLR4-dependent inflammatory response, promoting TLR4-mediated skin carcinogenesis [13]. But HMGB1 released from chemotherapy-induced dying cancer cells activates TLR4 expressed by DCs and induces antitumor T cell immunity [23]. Therefore, the ability of chemotherapeutic agents to kill tumors is decreased in TLR4- and MyD88-deficient mice. Moreover, breast cancer patients with an unfunctional TLR4 allele relapse more quickly than those carrying the normal TLR4 allele after radiotherapy and chemotherapy [23]. Endogenous HMGB1-mediated TLR2 activation on tumor-infiltrating myeloid DCs leads to TLR2-dependent brain tumor regression in mice [99]. The supernatants from the drugs-treated tumor cells specifically activate TLR2, and specific HMGB1 inhibitor or anti-HMGB1 antibodies can block TLR2 signaling and abolish therapeutic efficacy in a mouse model, highlighting the critical role of HMGB1-mediated TLR2 signaling to elicit tumor regression [99]. Actually, treatment of radiation or chemoagents results in release of HMGB1 from many other cancer cells such as melanoma, small cell lung carcinoma, and glioma cells, and therapeutic efficacy is positively correlated to the level of circulating HMGB1 [99].
Heat shock proteins (HSPs) are a class of endogenous TLR ligands. HSPs released from tumor cells can activate TAMs through TLR4. HSP60, HSP70, and HSP90 are expressed by melanoma cell K1735-M2 [14]. Pretreatment of K1735-M2 cells with neutralization antibodies against these molecules significantly reduces the production of TNF-α by WT macrophages that are cocultured with K1735-M2 cells. In contrast, K1735-M2 cells have no effects on TNF-α production by TLR4-deficient macrophages. HSP/TLR4-mediated secretion of growth factors by TAMs in turn promotes tumor growth via the NF-κB signaling pathway [14]. Thus, HSPs from tumor cells represent the endogenous ligands that dictate the TLR4-dependent inflammatory response in macrophages [14].
Two members of the S100 protein family, S100A8 and S100A9, have recently been identified as endogenous activators of TLR4 and have been shown to promote lethal, endotoxin-induced shock [100]. In addition to involvement in promoting the inflammatory response to infection, S100A8/S100A9 complex (also called calprotectin) is also associated with tumor development and spread [101, 102]. It has been demonstrated that the S100A8/S100A9 complex binds to colonic tumor cells, promoting NF-κB activation and cell proliferation [101]. It also enhances colitis-associated carcinogenesis in vivo [101]. Moreover, the growth of lymphomas and sarcomas in S100A9-deficient mice is significantly inhibited [102]. This effect is attributed to a reduced generation of myeloid-derived suppressor cells (MDSCs), which are immature myeloid cells that inhibit DC differentiation and lead to suppression of antitumor immune responses. Pancreatic adenocarcinoma up-regulated factor (PAUF) has been found to be a novel endogenous TLR2 and TLR4 ligand [103]. PAUF promotes metastasis by regulating TLR/CXCR4 activation. PAUF induces TLR2-mediated ERK activation to increase expression of the pro-tumorigenic cytokines RANTES and MIF in leukemia cells [103]. TLR4 also acts as a functional receptor for serum amyloid A (SAA) 3 in the pre-metastatic lungs, and SAA3 stimulates NF-κB signaling and facilitates metastasis [104]. In addition, small fragments of hyaluronan induce TLR4-dependent matrix metalloprotease- and cytokine-expression to promote melanoma invasiveness [105]. The extracellular matrix proteoglycan versican from Lewis lung carcinoma (LLC) strongly enhances LLC metastatic growth via TLR2/TLR6-mediated TNF-α secretion [106].
Angiogenesis is important for the development of tumor. The end products of lipid oxidation, ω-(2-carboxyethyl) pyrrole (CEP) as well as other related pyrroles, are generated during inflammation and wound healing and accumulate at high levels in highly vascularized tumors in both murine and human melanoma [107]. Carboxyalkylpyrroles-mediated TLR2 signaling promotes angiogenesis in hindlimb ischemia and wound healing models [107]. Neutralization of endogenous carboxyalkylpyrroles diminishes tumor angiogenesis. TLR2/MyD88 signaling is required for CEP-induced endothelial migration [107]. In addition, peroxiredoxin 1 (Prx1), an antioxidant and molecular chaperone, can control prostate cancer growth through TLR4-dependent regulation of tumor vasculature. Prx1 is overexpressed in human prostate cancer specimens. Inhibiting Prx1 expression in prostate tumor cells reduces tumor vascular formation and the levels of angiogenic proteins such as VEGF within the tumor microenvironment. Prx1 induces endothelial cell proliferation, migration, and differentiation in a TLR4-dependent manner [108]. These findings suggest that endogenous TLR signaling is involved in the angiogenesis during tumorigenesis [107, 108].
Which is the most valuable player
Infection and chronic inflammation are the key factors contributing to tumorigenesis. As we discussed above, both exogenous and endogenous TLR stimulators can trigger an inflammatory signaling, increasing neoplastic transformation of normal cells and regulating protumor or antitumor responses. Given that both types of molecules exist in the microenvironment of some tumors, such as those in the gastrointestinal tract, genital tract and skin that are exposed to microbes or microbial components delivered through blood circulation, the question is, which TLR ligand is the most valuable player (MVP) in tumorigenesis? Actually, to our best knowledge, this important issue remains under investigation. We believe that both exogenous and endogenous TLR ligands are equally important for tumor development and progression, but one of them may play a predominant role in a specific phase of a particular tumor type (Fig. 2). When microorganisms or their products are unavailable, the functional TLR signals are triggered only by endogenous ligands. As to infection-associated tumors, microbial components act as stimulatory molecules to sustain a TLR-mediated inflammatory response. This will benefit the elimination of pathogens and tissue repair during early phases of infection. But chronic inflammation and the ensuing release of endogenous molecules by inflamed cells promote cell transformation and carcinogenesis. Necrotic tumor cells further liberate endogenous TLR ligands, enhancing tumor cell proliferation and survival, and regulating the antitumor immune response.

Exogenous and endogenous TLR ligands are involved in the development of chronic inflammation-associated cancers. Microbial ligands activate TLRs and induce a chronic inflammation, resulting in tissue damage and cell transformation. Tumor cells and damaged cells/tissues release endogenous TLR ligands, which trigger TLRs expressed on tumor cells and immune cells such as dendritic cells and macrophages and regulate the progression or regression of tumors through the induction of cytokines and growth factors and modulation of antitumor immunity
Conclusion remarks
TLRs detect microbial infection and trigger an immune response to eliminate pathogens. In addition to microbial components, they respond to a great variety of host-derived molecules and regulate many pathological processes. Thus, both microbial and host-derived components contribute to the activation of TLRs. Much evidence indicates that TLR signaling is involved in tumorigenesis and associated with tumor development, progression, and prognosis.
Although a link between TLRs, inflammation, and carcinogenesis has been drawn, a number of questions still remain to be addressed. First, we still do not know exactly how much the TLR-mediated signaling contributes to the development of chronic inflammation-associated tumor. Understanding the underlying mechanism will facilitate the development of a new strategy for tumor therapy. Next, although accumulating evidence supports that both exogenous and endogenous TLR ligands play an important role in tumorigenesis, we do not know which is the major actor or the MVP. Current studies focus on the exogenous or endogenous TLR signaling independently, but their relationship or specific effects on tumorigenesis and tumor progression have not been investigated. Do they function sequentially or synergistically? Are TLRs able to discriminate exogenous ligands from endogenous ligands and respond differently? Moreover, according to previous studies, several endogenous TLR ligands are obviously implicated in tumor pathogenesis. But most of these host-derived molecules are the crucial mediators of non-infectious inflammation such as ischemia/reperfusion injury. Are there any tumor or tumor type-specific endogenous TLR ligands? Even the same ligand, such as HMGB1, can both promote tumor development while simultaneously activating the antitumor immunity. What is the underlying mechanism? In addition, we notice that the engagement of various TLRs may induce totally different biological effects. For example, TLR4 signaling usually promotes tumor growth, whereas TLR2 activation protects mice from tumor development. Why does different TLR signaling result in an opposite outcome? More attention should be paid to this. It should be mentioned here that some TLR ligands can also be recognized by other receptors except for TLRs. For example, the nucleotide binding oligomerization domain (NOD) receptors recognize specific motifs within the PGN. Poly(I:C) is recognized by TLR3 and cytosolic receptor RIG-I. HMGB1 can stimulate multiple receptors including receptor for advanced glycation end products (RAGE) and TLR4. Thus, comprehensive studies should be carried out to discriminate functions mediated by different receptors. We believe that all these important issues will be a frontier field of TLR study in the future. Understanding the role of exogenous and endogenous TLR ligands in tumorigenesis is of great value for tumor therapy.
References
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174:2942–2950
Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629
Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S (2004) A toll-like receptor that prevents infection by uropathogenic bacteria. Science 303:1522–1526
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782
Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F (1999) Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274:10689–10692
O’Neill LA, Bowie AG (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, Platt JL, Volk HD, Kupiec-Weglinski JW (2008) Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation 85:1016–1022
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22:5551–5560
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3:995–1001
Matzinger P (2002) The danger model: a renewed sense of self. Science 296:301–305
Mittal D, Saccheri F, Venereau E, Pusterla T, Bianchi ME, Rescigno M (2010) TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J 29:2242–2252
Lee CH, Wu CL, Shiau AL (2010) Toll-like receptor 4 signaling promotes tumor growth. J Immunother 33:73–82
Boraska Jelavic T, Barisic M, Drmic Hofman I, Boraska V, Vrdoljak E, Peruzovic M, Hozo I, Puljiz Z, Terzic J (2006) Microsatelite GT polymorphism in the toll-like receptor 2 is associated with colorectal cancer. Clin Genet 70:156–160
Pandey S, Mittal RD, Srivastava M, Srivastava K, Singh S, Srivastava S, Mittal B (2009) Impact of Toll-like receptors [TLR] 2 (−196 to −174 del) and TLR 4 (Asp299Gly, Thr399Ile) in cervical cancer susceptibility in North Indian women. Gynecol Oncol 114:501–505
He JF, Jia WH, Fan Q, Zhou XX, Qin HD, Shugart YY, Zeng YX (2007) Genetic polymorphisms of TLR3 are associated with Nasopharyngeal carcinoma risk in Cantonese population. BMC cancer 7:194
Nischalke HD, Coenen M, Berger C, Aldenhoff K, Muller T, Berg T, Kramer B, Korner C, Odenthal M, Schulze F, Grunhage F, Nattermann J, Sauerbruch T, Spengler U (2011) The toll-like receptor 2 (TLR2) −196 to −174 del/ins polymorphism affects viral loads and susceptibility to hepatocellular carcinoma in chronic hepatitis C. Int J Cancer doi:10.1002/ijc.26143. (Epub ahead of print)
Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA, Vaughan TL, McColl KE, Lissowska J, Zatonski W, Schoenberg JB, Blot WJ, Mowat NA, Fraumeni JF Jr, El-Omar EM (2007) A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 132:905–912
Ohara T, Morishita T, Suzuki H, Hibi T (2006) Heterozygous Thr 135 Ala polymorphism at leucine-rich repeat (LRR) in genomic DNA of toll-like receptor 4 in patients with poorly-differentiated gastric adenocarcinomas. Int J Mol Med 18:59–63
Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, Mayer L, Unkeless JC, Xiong H (2005) Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res 65:5009–5014
Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, Visintin I, Rutherford T, Mor G (2006) TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res 66:3859–3868
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059
He W, Liu Q, Wang L, Chen W, Li N, Cao X (2007) TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol 44:2850–2859
Tahara T, Arisawa T, Wang F, Shibata T, Nakamura M, Sakata M, Hirata I, Nakano H (2007) Toll-like receptor 2 −196 to 174 del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci 98:1790–1794
Srivastava K, Srivastava A, Kumar A, Mittal B (2010) Significant association between toll-like receptor gene polymorphisms and gallbladder cancer. Liver Int 30:1067–1072
Purdue MP, Lan Q, Wang SS, Kricker A, Menashe I, Zheng TZ, Hartge P, Grulich AE, Zhang Y, Morton LM, Vajdic CM, Holford TR, Severson RK, Leaderer BP, Cerhan JR, Yeager M, Cozen W, Jacobs K, Davis S, Rothman N, Chanock SJ, Chatterjee N, Armstrong BK (2009) A pooled investigation of Toll-like receptor gene variants and risk of non-Hodgkin lymphoma. Carcinogenesis 30:275–281
Nieters A, Beckmann L, Deeg E, Becker N (2006) Gene polymorphisms in Toll-like receptors, interleukin-10, and interleukin-10 receptor alpha and lymphoma risk. Genes Immun 7:615–624
Achyut BR, Ghoshal UC, Moorchung N, Mittal B (2007) Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum Immunol 68:901–907
Santini D, Angeletti S, Ruzzo A, Dicuonzo G, Galluzzo S, Vincenzi B, Calvieri A, Pizzagalli F, Graziano N, Ferraro E, Lorino G, Altomare A, Magnani M, Graziano F, Tonini G (2008) Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol 154:360–364
Rigoli L, Di Bella C, Fedele F, Procopio V, Amorini M, Lo Giudice G, Romeo P, Pugliatti F, Finocchiaro G, Luciano R, Caruso RA (2010) TLR4 and NOD2/CARD15 genetic polymorphisms and their possible role in gastric carcinogenesis. Anticancer Res 30:513–517
Hishida A, Matsuo K, Goto Y, Mitsuda Y, Hiraki A, Naito M, Wakai K, Tajima K, Hamajima N (2009) Toll-like receptor 4 +3725 G/C polymorphism, Helicobacter pylori seropositivity, and the risk of gastric atrophy and gastric cancer in Japanese. Helicobacter 14:47–53
Song J, Kim DY, Kim CS, Kim HJ, Lee DH, Lee HM, Ko W, Lee G (2009) The association between Toll-like receptor 4 (TLR4) polymorphisms and the risk of prostate cancer in Korean men. Cancer Genet Cytogenet 190:88–92
Chen YC, Giovannucci E, Lazarus R, Kraft P, Ketkar S, Hunter DJ (2005) Sequence variants of Toll-like receptor 4 and susceptibility to prostate cancer. Cancer Res 65:11771–11778
Zheng SL, Augustsson-Balter K, Chang B, Hedelin M, Li L, Adami HO, Bensen J, Li G, Johnasson JE, Turner AR, Adams TS, Meyers DA, Isaacs WB, Xu J, Gronberg H (2004) Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the CAncer Prostate in Sweden Study. Cancer Res 64:2918–2922
Cheng I, Plummer SJ, Casey G, Witte JS (2007) Toll-like receptor 4 genetic variation and advanced prostate cancer risk. Cancer Epidemiol Biomark Prev 16:352–355
Tahara T, Shibata T, Hirata I, Nakano H, Arisawa T (2009) CD14 promoter-159 polymorphism is associated with reduced risk of intestinal-type gastric cancer in a Japanese population. Dig Dis Sci 54:1508–1512
Ashton KA, Proietto A, Otton G, Symonds I, McEvoy M, Attia J, Scott RJ (2010) Toll-like receptor (TLR) and nucleosome-binding oligomerization domain (NOD) gene polymorphisms and endometrial cancer risk. BMC Cancer 10:382
Mollaki V, Georgiadis T, Tassidou A, Ioannou M, Daniil Z, Koutsokera A, Papathanassiou AA, Zintzaras E, Vassilopoulos G (2009) Polymorphisms and haplotypes in TLR9 and MYD88 are associated with the development of Hodgkin’s lymphoma: a candidate-gene association study. J Hum Genet 54:655–659
Castro FA, Forsti A, Buch S, Kalthoff H, Krauss C, Bauer M, Egberts J, Schniewind B, Broering DC, Schreiber S, Schmitt M, Hampe J, Hemminki K, Schafmayer C (2011) TLR-3 polymorphism is an independent prognostic marker for stage II colorectal cancer. Eur J Cancer 47:1203–1210
Zhou XX, Jia WH, Shen GP, Qin HD, Yu XJ, Chen LZ, Feng QS, Shugart YY, Zeng YX (2006) Sequence variants in toll-like receptor 10 are associated with nasopharyngeal carcinoma risk. Cancer Epidemiol Biomark Prev 15:862–866
Sun J, Wiklund F, Zheng SL, Chang B, Balter K, Li L, Johansson JE, Li G, Adami HO, Liu W, Tolin A, Turner AR, Meyers DA, Isaacs WB, Xu J, Gronberg H (2005) Sequence variants in Toll-like receptor gene cluster (TLR6-TLR1-TLR10) and prostate cancer risk. J Natl Cancer Inst 97:525–532
Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL (2009) Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res 69:3105–3113
Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M, Whiteside TL (2009) TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene 28:4353–4363
Yang H, Zhou H, Feng P, Zhou X, Wen H, Xie X, Shen H, Zhu X (2010) Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J Exp Clin Cancer Res 29:92
Hua D, Liu MY, Cheng ZD, Qin XJ, Zhang HM, Chen Y, Qin GJ, Liang G, Li JN, Han XF, Liu DX (2009) Small interfering RNA-directed targeting of Toll-like receptor 4 inhibits human prostate cancer cell invasion, survival, and tumorigenicity. Mol Immunol 46:2876–2884
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT (2007) Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133:1869–1881
Zhou B, Zhou H, Ling S, Guo D, Yan Y, Zhou F, Wu Y (2011) Activation of PAR2 or/and TLR4 promotes SW620 cell proliferation and migration via phosphorylation of ERK1/2. Oncol Rep 25:503–511
Carothers AM, Davids JS, Damas BC, Bertagnolli MM (2010) Persistent cyclooxygenase-2 inhibition downregulates NF-κB, resulting in chronic intestinal inflammation in the min/+ mouse model of colon tumorigenesis. Cancer Res 70:4433–4442
Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT (2006) Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology 131:862–877
Cammarota R, Bertolini V, Pennesi G, Bucci EO, Gottardi O, Garlanda C, Laghi L, Barberis MC, Sessa F, Noonan DM, Albini A (2010) The tumor microenvironment of colo-rectal cancer; stromal TLR-4 expression as a potential prognostic marker. J Transl Med 8:112
Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, Espana C, Ungaro R, Harpaz N, Cooper HS, Elson G, Kosco-Vilbois M, Zaias J, Perez MT, Mayer L, Vamadevan AS, Lira SA, Abreu MT (2011) Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis 17:1464–1473
Hernandez Y, Sotolongo J, Breglio K, Conduah D, Chen A, Xu R, Hsu D, Ungaro R, Hayes LA, Pastorini C, Abreu MT, Fukata M (2010) The role of prostaglandin E2 (PGE 2) in toll-like receptor 4 (TLR4)-mediated colitis-associated neoplasia. BMC Gastroenterol 10:82
Lowe EL, Crother TR, Rabizadeh S, Hu B, Wang H, Chen S, Shimada K, Wong MH, Michelsen KS, Arditi M (2010) Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PloS One 5:e13027
Ng LK, Rich AM, Hussaini HM, Thomson WM, Fisher AL, Horne LS, Seymour GJ (2011) Toll-like receptor 2 is present in the microenvironment of oral squamous cell carcinoma. Br J Cancer 104:460–463
Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, Ziparo E, Riccioli A (2008) Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC-alpha-dependent mechanism. Carcinogenesis 29:1334–1342
Matijevic T, Marjanovic M, Pavelic J (2009) Functionally active toll-like receptor 3 on human primary and metastatic cancer cells. Scand J Immunol 70:18–24
Nomi N, Kodama S, Suzuki M (2010) Toll-like receptor 3 signaling induces apoptosis in human head and neck cancer via survivin associated pathway. Oncol Rep 24:225–231
Chin AI, Miyahira AK, Covarrubias A, Teague J, Guo B, Dempsey PW, Cheng G (2010) Toll-like receptor 3-mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res 70:2595–2603
Conforti R, Ma Y, Morel Y, Paturel C, Terme M, Viaud S, Ryffel B, Ferrantini M, Uppaluri R, Schreiber R, Combadiere C, Chaput N, Andre F, Kroemer G, Zitvogel L (2010) Opposing effects of toll-like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res 70:490–500
Tanaka J, Sugimoto K, Shiraki K, Tameda M, Kusagawa S, Nojiri K, Beppu T, Yoneda K, Yamamoto N, Uchida K, Kojima T, Takei Y (2010) Functional cell surface expression of Toll-like receptor 9 promotes cell proliferation and survival in human hepatocellular carcinomas. Int J Oncol 37:805–814
Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, Chen D, Shackley B, Harris KW, Selander KS (2006) Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res 4:437–447
Ilvesaro JM, Merrell MA, Swain TM, Davidson J, Zayzafoon M, Harris KW, Selander KS (2007) Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate 67:774–781
Ren T, Wen ZK, Liu ZM, Liang YJ, Guo ZL, Xu L (2007) Functional expression of TLR9 is associated to the metastatic potential of human lung cancer cell: functional active role of TLR9 on tumor metastasis. Cancer Biol Ther 6:1704–1709
Xu L, Zhou Y, Liu Q, Luo JM, Qing M, Tang XY, Yao XS, Wang CH, Wen ZK (2009) CXCR4/SDF-1 pathway is crucial for TLR9 agonist enhanced metastasis of human lung cancer cell. Biochem Biophys Res Commun 382:571–576
Ilvesaro JM, Merrell MA, Li L, Wakchoure S, Graves D, Brooks S, Rahko E, Jukkola-Vuorinen A, Vuopala KS, Harris KW, Selander KS (2008) Toll-like receptor 9 mediates CpG oligonucleotide-induced cellular invasion. Mol Cancer Res 6:1534–1543
Di JM, Pang J, Sun QP, Zhang Y, Fang YQ, Liu XP, Zhou JH, Ruan XX, Gao X (2010) Toll-like receptor 9 agonists up-regulates the expression of cyclooxygenase-2 via activation of NF-κB in prostate cancer cells. Mol Biol Rep 37:1849–1855
Di JM, Pang J, Pu XY, Zhang Y, Liu XP, Fang YQ, Ruan XX, Gao X (2009) Toll-like receptor 9 agonists promote IL-8 and TGF-beta1 production via activation of nuclear factor kappaB in PC-3 cells. Cancer Genet Cytogenet 192:60–67
Brignole C, Marimpietri D, Di Paolo D, Perri P, Morandi F, Pastorino F, Zorzoli A, Pagnan G, Loi M, Caffa I, Erminio G, Haupt R, Gambini C, Pistoia V, Ponzoni M (2010) Therapeutic targeting of TLR9 inhibits cell growth and induces apoptosis in neuroblastoma. Cancer Res 70:9816–9826
Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, Arai A, Nakagawa Y, Suzuki K, Kitagawa M (2010) Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp Mol Pathol 88:293–298
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470:115–119
Yusuf N, Nasti TH, Long JA, Naseemuddin M, Lucas AP, Xu H, Elmets CA (2008) Protective role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res 68:615–622
Nicodemus CF, Wang L, Lucas J, Varghese B, Berek JS (2010) Toll-like receptor-3 as a target to enhance bioactivity of cancer immunotherapy. Am J Obstet Gynecol 202:e601–e608
Shojaei H, Oberg HH, Juricke M, Marischen L, Kunz M, Mundhenke C, Gieseler F, Kabelitz D, Wesch D (2009) Toll-like receptors 3 and 7 agonists enhance tumor cell lysis by human gammadelta T cells. Cancer Res 69:8710–8717
Rhee SH, Im E, Pothoulakis C (2008) Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology 135:518–528
Laplanche A, Alzieu L, Delozier T, Berlie J, Veyret C, Fargeot P, Luboinski M, Lacour J (2000) Polyadenylic-polyuridylic acid plus locoregional radiotherapy versus chemotherapy with CMF in operable breast cancer: a 14 year follow-up analysis of a randomized trial of the Federation Nationale des Centres de Lutte contre le Cancer (FNCLCC). Breast Cancer Res Treat 64:189–191
Jeung HC, Moon YW, Rha SY, Yoo NC, Roh JK, Noh SH, Min JS, Kim BS, Chung HC (2008) Phase III trial of adjuvant 5-fluorouracil and adriamycin versus 5-fluorouracil, adriamycin, and polyadenylic-polyuridylic acid (poly A:U) for locally advanced gastric cancer after curative surgery: final results of 15-year follow-up. Ann Oncol 19:520–526
Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF (2008) The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 13:859–875
Leonard JP, Link BK, Emmanouilides C, Gregory SA, Weisdorf D, Andrey J, Hainsworth J, Sparano JA, Tsai DE, Horning S, Krieg AM, Weiner GJ (2007) Phase I trial of toll-like receptor 9 agonist PF-3512676 with and following rituximab in patients with recurrent indolent and aggressive non Hodgkin’s lymphoma. Clin Cancer Res 13:6168–6174
Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, Kudo E, Shimada M, Sano T (2010) High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer 102:908–915
Gatti G, Quintar AA, Andreani V, Nicola JP, Maldonado CA, Masini-Repiso AM, Rivero VE, Maccioni M (2009) Expression of Toll-like receptor 4 in the prostate gland and its association with the severity of prostate cancer. Prostate 69:1387–1397
Yu L, Wang L, Li M, Zhong J, Wang Z, Chen S (2010) Expression of toll-like receptor 4 is down-regulated during progression of cervical neoplasia. Cancer Immunol Immunother 59:1021–1028
Vaisanen MR, Vaisanen T, Jukkola-Vuorinen A, Vuopala KS, Desmond R, Selander KS, Vaarala MH (2010) Expression of toll-like receptor-9 is increased in poorly differentiated prostate tumors. Prostate 70:817–824
Berger R, Fiegl H, Goebel G, Obexer P, Ausserlechner M, Doppler W, Hauser-Kronberger C, Reitsamer R, Egle D, Reimer D, Muller-Holzner E, Jones A, Widschwendter M (2010) Toll-like receptor 9 expression in breast and ovarian cancer is associated with poorly differentiated tumors. Cancer Sci 101:1059–1066
Xie W, Huang Y, Xie W, Guo A, Wu W (2010) Bacteria peptidoglycan promoted breast cancer cell invasiveness and adhesiveness by targeting toll-like receptor 2 in the cancer cells. PloS One 5:e10850
Grimm M, Kim M, Rosenwald A, Heemann U, Germer CT, Waaga-Gasser AM, Gasser M (2010) Toll-like receptor (TLR) 7 and TLR8 expression on CD133+ cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur J Cancer 46:2849–2857
Gonzalez-Reyes S, Fernandez JM, Gonzalez LO, Aguirre A, Suarez A, Gonzalez JM, Escaff S, Vizoso FJ (2011) Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol Immunother 60:217–226
Gonzalez-Reyes S, Marin L, Gonzalez L, Gonzalez LO, del Casar JM, Lamelas ML, Gonzalez-Quintana JM, Vizoso FJ (2010) Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer 10:665
Pei Z, Lin D, Song X, Li H, Yao H (2008) TLR4 signaling promotes the expression of VEGF and TGFbeta1 in human prostate epithelial PC3 cells induced by lipopolysaccharide. Cell Immunol 254:20–27
Rychahou PG, Kang J, Gulhati P, Doan HQ, Chen LA, Xiao SY, Chung DH, Evers BM (2008) Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci USA 105:20315–20320
Doan HQ, Bowen KA, Jackson LA, Evers BM (2009) Toll-like receptor 4 activation increases Akt phosphorylation in colon cancer cells. Anticancer Res 29:2473–2478
Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, Ferri LE (2011) LPS-Induced TLR4 Signaling in Human Colorectal Cancer Cells Increases beta1 Integrin-Mediated Cell Adhesion and Liver Metastasis. Cancer Res 71:1989–1998
Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, Tang L, Lin Y, He YQ, Zou SS, Wang C, Zhang HL, Cao GW, Wu MC, Wang HY (2010) Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology 52:1322–1333
Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, Mayer L, Unkeless J, Li D, Yuan Y, Zhang GM, Xiong H, Feng ZH (2007) Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res 67:4346–4352
Lu S, Lee J, Revelo M, Wang X, Lu S, Dong Z (2007) Smad3 is overexpressed in advanced human prostate cancer and necessary for progressive growth of prostate cancer cells in nude mice. Clin Cancer Res 13:5692–5702
Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54
Song EJ, Kang MJ, Kim YS, Kim SM, Lee SE, Kim CH, Kim DJ, Park JH (2011) Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int J Mol Med 28:115–119
Cai Z, Sanchez A, Shi Z, Zhang T, Liu M, Zhang D (2011) Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res 71:2466–2475
Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, Muhammad AK, Clark MC, Arditi M, Comin-Anduix B, Ribas A, Lowenstein PR, Castro MG (2009) HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 6:e10
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13:1042–1049
Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G (2008) RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 29:2035–2043
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249
Park HD, Lee Y, Oh YK, Jung JG, Park YW, Myung K, Kim KH, Koh SS, Lim DS (2011) Pancreatic adenocarcinoma upregulated factor promotes metastasis by regulating TLR/CXCR4 activation. Oncogene 30:201–211
Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y (2008) The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 10:1349–1355
Voelcker V, Gebhardt C, Averbeck M, Saalbach A, Wolf V, Weih F, Sleeman J, Anderegg U, Simon J (2008) Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp Dermatol 17:100–107
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, Luo JL, Karin M (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457:102–106
West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467:972–976
Riddell JR, Bshara W, Moser MT, Spernyak JA, Foster BA, Gollnick SO (2011) Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res 71:1637–1646
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ (2005) The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest 115:2223–2233
Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem 277:20847–20853
Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276:31332–31339
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277:15028–15034
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H (2002) HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 277:15107–15112
Zhang P, Cox CJ, Alvarez KM, Cunningham MW (2009) Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol 183:27–31
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11:1173–1179
Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R (2005) Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 52:2936–2946
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279:12542–12550
Chiron D, Bekeredjian-Ding I, Pellat-Deceunynck C, Bataille R, Jego G (2008) Toll-like receptors: lessons to learn from normal and malignant human B cells. Blood 112:2205–2213
Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, van den Berg WB, Radstake TR (2006) Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol 176:7021–7027
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW (2002) Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298:1025–1029
Smiley ST, King JA, Hancock WW (2001) Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol 167:2887–2894
Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF 3rd (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 276:10229–10233
Johnson GB, Brunn GJ, Kodaira Y, Platt JL (2002) Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol 168:5233–5239
Ohashi K, Burkart V, Flohe S, Kolb H (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 164:558–561
Chase MA, Wheeler DS, Lierl KM, Hughes VS, Wong HR, Page K (2007) Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-κB-dependent mechanism. J Immunol 179:6318–6324
Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249
Riddell JR, Wang XY, Minderman H, Gollnick SO (2010) Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol 184:1022–1030
Tarkowski A, Bjersing J, Shestakov A, Bokarewa MI (2010) Resistin competes with lipopolysaccharide for binding to Toll-like receptor 4. J Cell Mol Med 14:1419–1431
Guillot L, Balloy V, McCormack FX, Golenbock DT, Chignard M, Si-Tahar M (2002) Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol 168:5989–5992
Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B (2009) Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15:774–780
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A (2005) RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med 202:1171–1177
Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med 11:263–270
Sioud M (2005) Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J Mol Biol 348:1079–1090
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A (2002) Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416:603–607
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A (2003) Activation of autoreactive B cells by CpG dsDNA. Immunity 19:837–847
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No. 31070733 and No. 81172485), the PhD Program Foundation of Ministry of Education of China (No. 20090171110029), and the National Basic Research Program of China (No. 2011CB946101).
Conflict of interest statement
The authors state no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, L., Wang, L. & Chen, S. Exogenous or endogenous Toll-like receptor ligands: which is the MVP in tumorigenesis?. Cell. Mol. Life Sci. 69, 935–949 (2012). https://doi.org/10.1007/s00018-011-0864-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0864-6