
Abstract
Purpose
In spite of the recent advances in surgery and antitumor drugs, the brain tumors, like glioblastoma, have shown a poor prognosis. The aim of this study was to determine the effect of pertussis toxin (PTx) as immunomodulatory molecule on glial tumors induced by C6 glioma cells.
Methods
Given the pleiotropic effect of PTx on the immune system, we analyzed the effect of PTx on CD4+/CD25+/FoxP3+ (Treg) cells like as immunotherapeutic adjuvant. Thirty rats with a glial tumor of 1.5 cm in diameter were separated in two groups: the first group was treated with PTx and the second group was non-treated (controls). Tumoral volume was measured weekly; tumor, blood and spleen were taken for analysis of subpopulations of T cells, apoptotic index and cytokine contents, in both groups.
Results
We observed a significant decrease in tumor volume in the PTx group; this was associated with a decreased in the number of Treg cells, in both spleen and tumor. The percentage of apoptotic cells was increased as compared with that of controls. The production of proinflammatory cytokines was increased in mRNA for IL-6 as well as a small increase in the mRNA expression of perforin and granzime in tumors from rats treated with PTx. No changes were found in the mRNA expression of MCP-1 and MIP-1α.
Conclusion
These results suggest that PTx could be an immunotherapeutic adjuvant in the integral therapy against glial tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant gliomas are the most frequent brain tumors in adults, and they are responsible for more than 15,000 deaths in the United States each year (Deorah et al. 2006). At the National Institute of Neurology and Neurosurgery of Mexico, Glioblastoma multiforme (GBM) represents 9% of all brain tumors and 45.7% of primary gliomas (Lopez-Gonzalez and Sotelo 2000; Velasquez-Perez and Jimenez-Marcial 2003). Despite the multiple advances in diagnosis, the prognosis for this tumor is poor; the median survival time of untreated tumors is about 5 months. Even with the best available current therapy, which includes radiation, chemotherapy and surgery, the median survival does not extend beyond 14 months (Buckner 2003; Stupp et al. 2005).
In other way, it has been shown that the progression of some malignant tumors is associated with the expression of tumor-specific antigens and with antigen-specific immune response (Jager et al. 2001). Hence, theoretically, effective tumor rejection and immunity could be achieved by immunization with tumor-associated antigens. However, active immunotherapy for cancer has shown only minimal clinical effectiveness due to the mechanism of evasion in the immune system.
Regulatory T cells, also known as CD4+CD25+FoxP3+, Treg, are a subpopulation of T cells that inhibits the activation of the immune response, thereby maintaining the homeostasis of the immune system and tolerance to self-antigens. The depletion of Treg’s seems necessary to achieve an effective immune response (Garcia-Lora et al. 2003). There is increasing evidence for the existence of elevated numbers of regulatory T cells in solid tumors and hematological malignancies (Barnett et al. 2005; Chattopadhyay et al. 2005; Linehan and Goedegebuure 2005). The presence of infiltrating Treg may be detrimental to the host natural defences against the tumor; while the presence of effector T lymphocytes, including CD8+ and non-regulatory CD4+ helper T cells, may be beneficial (Fecci et al. 2006). Lesniak and col. recently have shown an increased number of Treg both, in tumor-infiltrating lymphocytes as well as in peripheral blood of patients with GBM. Depletion of Treg increased survival of mice with experimental brain tumors (El Andaloussi et al. 2006; El Andaloussi and Lesniak 2006).
Pertussis toxin (PTx) has been used for a long time as adjuvant to enhance the severity of experimental autoimmune encephalomyelitis (EAE) in permissive strains and even to render resistant strains susceptible (Linthicum 1982; Munoz et al. 1984). Its properties are not restricted to CNS autoimmunity, but also apply to other systemic models of autoimmunity such as orchitis, uveitis and inflammatory myopathy (Agarwal et al. 2002; Hart et al. 1987; Hou et al. 2003; Kohno et al. 1983). PTx is highly immunogenic (Mascart et al. 2003; Nencioni et al. 1991); therefore, it has been included in some vaccines in a chemically or genetically detoxified form (Tonon et al. 2002). Numerous immunological effects have been attributed to PTx. On the innate immune system, PTx decreases the production of IL-6 and IL-10 by mast cells (Mielcarek et al. 2001), promotes maturation of APC leading to the upregulation of MHC class II or co-stimulatory molecules, and incites the production of IL-12 (Darabi et al. 2004; He et al. 2000; Hou et al. 2003; Tonon et al. 2002). On the adaptive immune system, PTx increases both Th1 and Th2 responses (Hofstetter et al. 2002; Ryan et al. 1998; Shive et al. 2000) and inhibits chemokine-induced lymphocyte migration (Alt et al. 2002; Cyster and Goodnow 1995). Classically, the exacerbating effect of PTx on EAE was attributed to increased sensitization to histamine (Linthicum 1982; Linthicum and Frelinger 1982) and permeabilization of the blood–brain barrier (Bruckener et al. 2003). More recently, PTx has shown a selective effect over the Treg cell population, inducing a remarkable reduction in the frequency and immunosuppressive activity of splenic CD4+CD25+T cells in vivo (Cassan et al. 2006; Chen et al. 2006). The aim of the present study was to determine the effect of PTx as immunomodulatory molecule on malignant glial tumors induced by C6 glioma cells.
Methods
C6 glioma in rats
The animal model protocol here used was approved by the animal research and care committee of the Instituto Nacional de Neurologia y Neurocirugia. Rat C6 glioma cells (Benda et al. 1968), obtained from the American Type Culture Collection (Rockville, MD), were cultured under sterile conditions at 37°C in a humid environment with 5% of CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS). The technique for achieving reproducible C6 gliomas has been previously reported elsewhere (Arrieta et al. 1998; Pineda et al. 2005). Briefly, 1 × 107 cultured C6 cells were intraperitoneally inoculated into 12-week-old female Wistar rats. On twentieth day of post-inoculation, a large peritoneal tumor develops (Guevara and Sotelo 1999). This tumor was crushed and mechanically dissociated by successive passage through 14, 16, 18 and 21 gauge needles at 4°C. A suspension of 1 × 107 viable cells in 1 ml of saline solution was subcutaneously inoculated into the left thigh of 12-week-old female Wistar rats. Approximately 7 days after cell implantation, subcutaneous tumors were visible in 80% of the animals; these tumors reached a diameter of 1.5 cm in three weeks; not spontaneous involution was observed of tumors at that size. Therefore, only rats that developed subcutaneous C6 gliomas of at least 1.5 cm in diameter were included in the study.
Experimental design
Thirty rats with tumor of 1.5 cm diameter were separated in two groups: the first was treated with vehicle alone (control group) and the second group was treated with PTx. Tumor growth was assessed once a week by size measurement with a caliper. Animals were euthanized on day 21. Tumor, blood and spleen of all animals grown both groups were taken for analysis.
To induce the pleiotropic effects on the immune system by PTx, we injected Wistar rats intraperitoneally with 2 μg PTx at day 0, and 1 μg at day 2, as described by Cassan et al. (2006). Control rats only received PBS with the same schedule.
Determination of tumoral volume
Tumors were measured weekly and their volumes (in cubic centimeters) were determined for each rat with the formula 6π × L × W × H, as described by Tomayko and Reynolds (1989). After 7, 14, 21 and 28 days of treatment, five animals per group were anaesthetized and killed by exsanguination. Blood, spleen and tumor samples were taken for further determinations. In accordance with our institutional guidelines for the compassionate use of experimental animals, rats in which the tumor reached a volume of more than 50 cm3 before the end of the experiment were euthanized.
Flow cytometry of T-lymphocyte subpopulations
Direct immunofluorescence using monoclonal antibodies anti-CD4-PE, anti-CD8-PE, anti-CD56-FITC, anti-CD25-APC (Biosource, Washington, USA) and anti-Foxp3-PE (e-biosciences, San Diego, CA) were used to determine the percentages of CD4+, CD8+, CD56+ and CD4+/CD25+/FoxP3+ (Treg) lymphocytes in the peripheral blood, spleen and tumor samples. Briefly, 30 μl of either blood or tumor or spleen homogenates was incubated 30 min with 5 μl of the corresponding monoclonal antibody (1:100 dilutions). Afterward, 200 μl of red blood lysis solution (BD Biosciences, Palo Alto, CA) was added, incubated for 10 min and washed twice with 0.1% BSA and 0.1% NaN3 PBS (pH 7.2). Afterward, 200 μl of permeabilizing solution was added and the cells were incubated for 10 min, washed and further incubated with anti-FoxP3-APC for 30 min, washed and fixed in 1% paraformaldehyde/PBS for examination by flow Cytometry (FACSCalibur, BD Biosciences) Cell Quest Pro software (BD Biosciences); 10,000 events in the region corresponding to lymphocytes were analyzed. From this region, the percentage of positive cells from each sample was determined. Results were expressed as the percentage of fluorescent positive cells (±SD).
Immunohistological analysis
Ten tumors from each group were extracted and included in paraffin for immunohistochemical analyses. Thin sections (5 μm) were stained with anti-CD4 monoclonal antibodies coupled to allophycocyanin (APC) (Biosource, Washington, USA) monoclonal antibodies. Additionally, five tumors of each group were stained with anti-CD68 monoclonal antibodies coupled to fluorescein (FITC) (Santa Cruz Biotechnology California, USA), and 5 fields were counted of each slide. Cell nuclei were stained with propidium iodide (PI) for fluorescence microscopic analysis.
Determination of apoptosis use Sub-G 0 peak method
At the end of the experiment, the rats were killed and the tumor was removed, minced into 1–2 mm fragments and pressed through a wire mesh. Resulting cells were suspended on 1 ml of PBS, filtered and adjusted to a concentration of 1 × 106 cell/ml; these suspensions were fixed in cold ethanol/water (70%) and stored at −20°C until analysis. The cells were suspended on PBS, centrifuged and resuspended in 0.5 ml PBS; 250 μl of citrate buffer was added and incubated 7 min at 37°C. Then, the cells were centrifuged and the pellet was resuspended in propidium iodine solution (20 μg/ml) (a fluorescent vital dye that stains DNA), and RNAse (5 mg/ml). Propidium iodide does not cross the plasma membrane of cells that are either viable or in the early stages of apoptosis because they maintain membrane integrity; in contrast, the cells in the late stages of apoptosis or already dead have lost membrane integrity and are permeable to propidium iodide; the cells were incubated in the dark for 30 min at room temperature and kept in the dark at 4°C for 20 min. The fluorescence of individual nuclei was measured using a FACSCalibur flow cytometer (Becton–Dickinson). Measurements were taken at 488 nm, gating out doublets and clumps for each sample, and the 10,000 events in the gate were evaluated.
Detection of apoptosis by Annexin V-7AAD
At the end of the experiment, the remaining rats were killed, and the tumor was removed, minced into 1–2 mm fragments and pressed through a wire mesh. The cells were suspended on 1 ml of PBS, filtered and adjusted to a concentration of 1 × 106 cell/ml; these suspensions were stained with Annexin V-7 AAD kit (PE Annexin V Apoptosis Detection Kit I, BD Pharmingen) for 15 min at room temperature. Cells were washed, resuspended in FACS buffer and analyzed by cytometry within 1 h. Data were collected on a FACSCalibur instrument (BD Biosciences), and analysis was performed with the CellQuest Pro software (BD Biosciences).
TUNEL staining
Ten tumors from each group were extracted and included in paraffin for immunohistochemical analyses. Thin sections (5 μm) were stained for terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) using an Apo-BrdU-Red in situ DNA Fragmentation Assay Kit (Biovision) according to the manufacturer’s instructions.
Toxicity studies on hematic and biochemical parameters
For studies of blood count and blood chemistry (glucose, BUN, creatinine and liver function tests), five rats from each group were anaesthetized and blood samples were obtained by intracardiac puncture. The same parameters, determined in five healthy rats, were taken as control values.
Gene expression analysis by real-time PCR
Total RNA was extracted from rat tumor samples at the indicated time points after tumor implants. The RNA extraction was made by using the TRIZOL® Reagent (Gibco BRL) as indicated by the manufacturer. To minimize the risk of contaminating DNA, total RNAs were digested with 10 U of DNase I (Boehringer Mannheim, Lewes, UK) for 15 min at 37°C, and followed by phenol–chloroform extraction and re-precipitated in 2 volumes of absolute ethanol and 0.5 volume of 3 M sodium acetate (Pineda et al. 2005), cDNAs for RT-PCR were synthesized from 300 ng of total RNA using 1 μM oligo (dT12), at 65°C for 10 min. The RT mix, consisting of 1× buffer, 2 U of Moloney murine leukemia virus reverse transcriptase (Life Technology, Paisley, UK), 20 μM of deoxynucleoside triphosphates (dNTP) (Pharmacia Biotech, Herts, UK) and 10 mM dithiothreitol (Life technology), was added, and this mixture was incubated at 37°C for 1 h.
The primers used to amplify FOXP3 were the following: FOXP3F: CCTGTTCCTTCTCATCACTGGC, and FOXP3R: GCTTTTAGCCTGAACCCCCTTA; IL-6F: CTT CTT GGG ACT GAT GTT GTT GA and IL-6R GGA AGT TGG GGT AGG AAG GAC; MIP-1αF: AGC TTG ACG GTG ACC CCT CCA G and MIP-1αR: GGT CAG TTA GCC TTG CCT TTG TTC; MCP-1F: CGC TTC TTG GCC TGT TGT T and MCP-1R: CTG CTG CTG CTG ATT CTC TTG TAG; PerforinF: GAAACAACACAAAATCGCCACC and PerforinR: TTCCCGAAGAGCAGGTCATTC; and Granzyme BF: CTGTGGTGAAAATCATTCCCCA and Granzyme BR: AGCGCTAGACCTCTTGGCCTTA. Primers were purchased from Invitrogen.
Quantitative analysis of cDNA amplification was assessed by incorporation of SYBR Green into cDNAs. PCRs containing 1 μg of cDNA template, 0.5 μM of each primer set and SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) were performed in a total volume of 25 μl under the following conditions: one cycle of 10 min at 95°C, 40 cycles of 10 s at 95°C and 1 min at 60°C. Gene expression was analyzed using the ABI Prism 7500 sequence detector (Applied Biosystems) and ABI Prism SDS software version 1.9.1 (Applied Biosystems). Data were normalized by dividing the quantity of the FOXP3 gene by the expression value of the endogenous reference gene (GADPH from Applied Biosystems). Triplicate reactions were performed using a pool of all cDNA samples of each time point. Relative expression was calculated for each gene by using the ΔCT (threshold cycle) method.
Analysis of pertussis toxin-mediated toxicity in vitro
For determination of the cytotoxic effect mediated by our treatment, C6 cells were grown by triplicate in 6-well plates (3 × 105 cells per well) for 24 h and subsequently incubated with different concentrations of PTx (from 0 to 1,000 ng/ml) for 24 h. Apoptosis induction was measured by using Annexin V-7AAD as described above.
Statistical analysis
Groups were compared by the Student’s t-tests; P value less than 0.05 was defined as significant. All statistical analyses were performed with the SPSS statistical software package (SPSS 12.0 for Windows; SPSS Inc., Chicago, IL).
Results
Antitumoral effect of PTx on rats bearing subcutaneous C6 glioma
All animals survived until the end of the experiment. In all controls, the tumor grew to a very large size (over 40 cm3); there was no case of spontaneous involution. Rats treated with PTx exhibited 77% tumoral reduction (mean volume, 8.7 ± 3.1) as compared with controls (mean volume, 39.75 ± 5.7 (P = 0.04) (Fig. 1).

Effect of PTx treatment on tumor growth. Rats were inoculated with C6 cells until the tumor grew (around 2 cm diameter). Treatment consisted of vehicle alone a in controls (circles) or b PTx 2 μg at day 0 and 1 μg at day 2 (squares). Tumor volume was estimated using the standard formula π/6 × length × width2. Results are expressed as means ± SEM of tumor volume measured in 10 rats per group
Changes of CD4+CD25+ FoxP3 T-cell population after PTx treatment
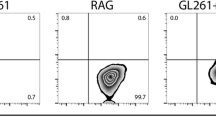
No differences were found between groups in the total number of splenocytes or in the percentage of CD4+, CD8+ and CD56+ splenocytes (Fig. 2c–e) after PTx treatment. In contrast, PTx treatment decreased Treg cells in the spleen and increased them in blood: A significant decrease in 83% of CD4+ splenocytes expressing CD25 and FoxP3 regulatory T cells (Treg) was seen in PTx-treated rats (P = 0.03) (Fig. 2f). In blood (Fig. 3), no significant changes in CD4+ (Fig. 3c), CD8+ (Fig. 3d) and CD56+ (Fig. 3e) lymphocytes were seen in rats treated with PTx as compared with controls, whereas significant increase in the number of Treg was seen in the PTx-treated group as compared with controls (P = 0.04) (Fig. 3f).

Comparative study of the characterization of c CD4+, d CD8+, e CD56+ and f Treg splenocytes by surface staining of CD4, CD8, CD56 and CD25 or by FoxP3 intracellular staining in rats with a C6 glioma, a and b are representative dot plots of control and PTx group, respectively. Treatment consisted of vehicle alone (left column) or PTx 2 μg at day 0 and 1 μg at day 2 (right column). Animals were killed on day 21 after treatment. Isolated splenocytes were stained with anti-CD4, anti-CD8, anti-CD56, anti-CD25 and FoxP3 mAbs and submitted to flow cytometry analysis. Results are expressed as mean ± SD of cells percentage in 10 rats of each group

Comparative study of the characterization of c CD4+, d CD8+, e CD56+ and f Treg cells in peripheral blood by surface staining of CD4, CD8, CD56 and CD25 or by FoxP3 intracellular staining in rats with a C6 glioma, a and b are representative dot plots of control and PTx group respectively. Treatment consisted of vehicle alone (left column) or PTx 2 μg at day 0 and 1 μg at day 2 (right column). Animals were killed on day 21 by exsanguination. Peripheral blood cells were stained with anti-CD4, anti-CD8, anti-CD56 and FoxP3 mAbs and then submitted to flow cytometry analysis. Results are expressed as mean ± SD of cell percentage in 10 rats of each group
PTx treatment decrease CD4+ cells and Treg’s lymphocytes within the tumors
Immunofluorescence analysis of CD4+ tumor-infiltrating lymphocytes (TIL) demonstrated a large number of CD4+ cells in the tumor of control rats group (Fig. 4a) and reduction in these cells in the tumor of rats treated with PTx (Fig. 4b). Analysis of TIL did not show significant decrease in the tumor of rats treated with PTx changes in the percentage of CD4+ (Fig. 4c) or CD8+ cells (Fig. 4d), whereas there was a significant decrease (P < 0.04) in the percentage of CD56+ (Fig. 4e) and Treg cells (P = 0.03) (Fig. 4f).

Determination of CD4+ inside a C6 glioma in rats by immunofluorescence (a, b) or CD4+, CD8+, CD56+ and Treg’s by FACS (c, d, e, f). Treatment consisted of vehicle (left column) or PTx 2 μg at day 0 and 1 μg at day 2 (right column). Animals were killed on day 21, and tumors were stained with antibodies anti-CD4 coupled to APC and the nuclei was counterstained with ethidium bromide for immunofluorescence. To the FACS analysis 1 cm3 of the each tumor was mechanically minced and cells were stained with anti-CD4, anti-CD8, anti-CD56, and FoxP3 mAbs and then analyzed by flow cytometry. Results are expressed as mean ± SD of the percentage of CD4+, CD8+, CD56+ and Treg’s from 10 rats in both groups
Treatment with PTx decreases the viability and induces apoptosis of tumor cells
Immunofluorescence analysis of TUNEL-positive cells (Fig. 5a, b) shown an increase in the number of TUNEL-positive cells in tumors treated with PTx (Fig. 5b) as compared with controls (Fig. 5a). A significant decrease (P = 0.04) in the percentage of live cells was seen in the tumor of rats treated with PTx (Fig. 5c). The percentage of apoptotic tumoral cells in the PTx group was increased (P = 0.01) as compared with controls (Fig. 5d). Similar results were seen by the sub-Go peak method (Fig. 5e) with a significant increase in apoptosis in the PTx group (P = 0.007). In the TUNEL assay, the number of apoptotic cells in the PTx group largely increased as compared with controls (Fig. 5a, b, f) (P < 0.001).

Determination of apoptosis by TUNEL staining inside a C6 glioma in a controls or b PTx treated c percentage of viability of tumors from 10 rats with C6 glioma. Treatment consisted of vehicle (left column) or PTx 2 μg at day 0 and 1 μg at day 2 (right column). Percentage of viability was assed; 1 cm3 of the each tumor was mechanically minced and stained with blue trypan cells were counted in a Neubauer chamber. d Apoptosis was measured by Annexin V-PE/7AAD by FACS. e Sub-G0 peak was measured from around 1 cm3 of minced tumor and analyzed by FACS and E number of TUNEL-positive cells counted in ten fields, graphs shows the mean ± SD from 10 rats in both groups
PTx toxicity on hematological parameters
All animals survived until the end of the experiment. In controls, the tumor grew to a very large size (over 40 cm3); no case of spontaneous involution was seen. Comparisons of hematological and chemical blood parameters measured at the end of the study did not show differences between groups.
Gene expression analysis by real-time RT-PCR
A small increment in mRNA expression of IL-6 (3.74 fold higher), MIP-1α (1.7 fold higher), granzyme (1.5 fold higher) and perforin (0.9 fold higher) was observed in tumors from rats treated with PTx, as compared with controls (the value for controls was 1 for all comparisons) (Fig. 6).

Transcripts for a IL-6, b MCP1-alpha, c MIP-1alpha d FoxP3, e granzime and f perforine in the tumor of rats treated with vehicle alone, n = 10 (left column) or PTx 2 μg at day 0 and 1 μg at day 2, n = 10 (right column). All the samples were normalized to GADPH = glyceraldehyde 3-phosphate dehydrogenase graphs, shows the mean ± SD from 10 rats in both groups
Toxicity of PTx in vitro
C6 glioma cells exposed to different concentrations of PTx (from 0 to 1,000 ng/ml) for 24 h, stained with Annexin V-7 AAD, showed small increase in the percentage of apoptotic cells after 20 ng/ml (from 6 to 12%); this increase continued for higher concentrations (200 and 1,000 ng/ml) (Fig. 7).

In vitro effect of a PTx treatment on C6 glioma cells. 3 × 105 cells were exposed to different concentrations of PTx (0, 2, 20, 200 and 1,000 ng/ml), and the percentage of apoptotic cells were determined by FACS. Increase in the percentage of apoptotic cells was seen in cells treated with 20, 200 and 1,000 ng/ml of toxin; no changes were seen in cells treated with 2 ng/ml of PTx as compared with non-treated cells
PTx treatment decrease CD68+ glioma-invading macrophages
Immunofluorescence analysis of CD68+ glioma-invading macrophages (GIM) demonstrated a large number of CD68+ cells in the tumor of control rats group (Fig. 8a) and reduction in these cells in the tumor of rats treated with PTx (Fig. 8b). Analysis of GIM did show a significant decrease in the number of CD68+ cells in the PTx group when it was compared with controls (P < 0.001) (Fig. 8c).

Immunofluorescence analysis of macrophages (CD68+). a Tumor of control rats with a high number of glioma-invading macrophages. b Effect of PTx treatment on glioma showing a diminished number of macrophages. c The graphic shows the differences in quantification in both, control and PTx treatment. Results are expressed as mean ± SD of cell number in five fields in 5 rats of each group
Discussion
Patients with malignant glioma have a markedly increased Treg fraction in peripheral blood and within the tumor, which might block cellular immune responses against the tumor (Fecci et al. 2006). Large numbers of Treg cells are present in the blood of patients with a variety of neoplasms, including malignant glioma; these cells play a significant role in suppression of antitumor immunity (Curiel et al. 2004; Ichihara et al. 2003; Liyanage et al. 2002; Woo et al. 2001). In patients with malignant glioma, such increase in Treg cells in peripheral blood correlate with local immune depression typical in these patients, e.g., impaired T-cell proliferative responses and counterproductive shifts toward TH2 cytokine production (Fecci et al. 2006). Due to the relevant participation of Treg cells in the immune response, their modulation could improve endogenous protection against glioma cells. In this study, we use PTx as immunomodulatory treatment in C6 glioma model to explore its potential capacity to reduce Treg’s in spleen, blood and Treg’s TIL. C6 glioma model has been widely used by our group and others for experimental therapy (Guevara and Sotelo 1999; Zhu et al. 2005; Li et al. 2011), the subcutaneous implantation of C6 in young rats leads to a high rate of tumor development; after the tumor reaches 1.5 cm in diameter, approximately at day 20, no spontaneous involution occurs. Therefore, in this study, animals that developed a tumor of this diameter were selected for the study.
Rats treated with PTx exhibited a tumoral reduction of 77% as compared with controls. PTx caused specific depletion of Treg subpopulations from the spleen without parallel changes in CD4+, CD8 or CD56+ lymphocytes (Cassan et al. 2006; Chen et al. 2006), The small increase in the percentage of Treg in peripheral blood from rats treated with PTx may be due to a compensatory effect of the Treg depletion induced in the spleen; the decrease in Treg cells was seen mostly in the spleen and to a lesser extend in bone marrow, whereas the liver was not affected (Cassan et al. 2006; Chen et al. 2006). Our results, similar to other reports, show that Treg cells are increased in glioma infiltrates and correlated directly with tumoral growth (El Andaloussi et al. 2006; El Andaloussi and Lesniak 2007). Our results show that the treatment with PTx decreases the percentage and number of Treg’s infiltrating the tumor. Moreover, the PTx treatment had a deleterious effect on tumoral growth with decrements in C6 cell viability and apoptosis increase in glioma cells. Although PTx had a direct effect on the induction of apoptosis of C6 glioma cells, the percentage of apoptotic cells was not sufficient to eliminate the tumor. Treg cells with suppressive phenotypes in gliomas downregulate antitumor response within the CNS (El Andaloussi et al. 2006); conversely, depletion of Treg in vivo by anti-CD25 mAb induces the rejection of various immunogenic tumors in mice (Golgher et al. 2002; Onizuka et al. 1999; Shimizu et al. 1999). The induction of apoptosis could be a direct effect of the immune response against tumor cells. Chen et al. (2007) demonstrated that the combination of IL-1β, IL-6 and TNF or the use of adjuvants that promote these cytokines results in a robust proliferation of T effectors cells despite the presence of Treg cells, providing a novel interpretation for the adjuvant-like action of pro-inflammatory cytokines; the combination of these cytokines inhibits the function of Treg. In our study, the treatment with PTx induced a 2.5-fold increase in mRNA for IL-6 accompanied by a remarkable reduction, without increments in the expression of MCP-1α and MIP-1α, in the expression of FoxP3 mRNA. Our results suggest that PTx treatment could promote the generation of T effector cells to IL17 inside the tumor, as a consequence of an adaptive immune response against tumor cells (Chen et al. 2007). Additionally, as NK cells could participate in the control of tumors, the increased expression of perforin and grazyme suggests the possibility that NK response against tumor cells could also be involved in the observed reduction in tumor size in rats treated with PTx.
On the other hand, Backlund et al. (1985) and Papaspyridonos et al. (2008) demonstrated that the PTx treatment inhibits the chemotaxis of macrophages (RAW264 and WBC264-9C cell lines) in vitro (Backlund et al. 1985; Papaspyridonos et al. 2008). Increase in the number of CD68 has been associated with high-grade tumors and low rates in survival (Strojnik et al. 2009). We found that the number of CD68 was diminished in the group treated with PTx. It is possible that treatment with PTx reduces invasion, and metastasis of one tumor type would likely be effective in treating tumors with tumor-invading macrophages (TIM). The dual effect of the PTx over the inhibition of macrophages chemotaxis and selective depletion of Tregs within the tumor could warrant the use of PTx in tumors with these infiltrative characteristics. Overall, the presented data support earlier findings that PTx attenuates in vivo tumor growth by inducing cell apoptosis of C6 glioma cells.
The PTx treatment and pro-inflammatory effect of the C6 cells could induce an autoimmune response against the tumor. Our results warrant further studies of immunotherapy with PTx as potential adjuvant to the conventional treatment against malignant glioma.
References
Agarwal RK, Sun SH, Su SB, Chan CC, Caspi RR (2002) Pertussis toxin alters the innate and the adaptive immune responses in a pertussis-dependent model of autoimmunity. J Neuroimmunol 129:133–140
Alt C, Laschinger M, Engelhardt B (2002) Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol 32:2133–2144
Arrieta O, Guevara P, Reyes S, Ortiz A, Rembao D, Sotelo J (1998) Protamine inhibits angiogenesis and growth of C6 rat glioma; a synergistic effect when combined with carmustine. Eur J Cancer 34:2101–2106
Backlund PS Jr, Meade BD, Manclark CR, Cantoni GL, Aksamit RR (1985) Pertussis toxin inhibition of chemotaxis and the ADP-ribosylation of a membrane protein in a human-mouse hybrid cell line. Proc Natl Acad Sci USA 82:2637–2641
Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ (2005) Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol 54:369–377
Benda P, Lightbody J, Sato G, Levine L, Sweet W (1968) Differentiated rat glial cell strain in tissue culture. Science 161:370–371
Bruckener KE, el Baya A, Galla HJ, Schmidt MA (2003) Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J Cell Sci 116:1837–1846
Buckner JC (2003) Factors influencing survival in high-grade gliomas. Semin Oncol 30:10–14
Cassan C, Piaggio E, Zappulla JP, Mars LT, Couturier N, Bucciarelli F, Desbois S, Bauer J, Gonzalez-Dunia D, Liblau RS (2006) Pertussis toxin reduces the number of splenic Foxp3+ regulatory T cells. J Immunol 177:1552–1560
Chattopadhyay S, Chakraborty NG, Mukherji B (2005) Regulatory T cells and tumor immunity. Cancer Immunol Immunother 54:1153–1161
Chen X, Winkler-Pickett RT, Carbonetti NH, Ortaldo JR, Oppenheim JJ, Howard OM (2006) Pertussis toxin as an adjuvant suppresses the number and function of CD4+CD25+ T regulatory cells. Eur J Immunol 36:671–680
Chen X, Howard OM, Oppenheim JJ (2007) Pertussis toxin by inducing IL-6 promotes the generation of IL-17-producing CD4 cells. J Immunol 178:6123–6129
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Cyster JG, Goodnow CC (1995) Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J Exp Med 182:581–586
Darabi K, Karulin AY, Boehm BO, Hofstetter HH, Fabry Z, LaManna JC, Chavez JC, Tary-Lehmann M, Lehmann PV (2004) The third signal in T cell-mediated autoimmune disease? J Immunol 173:92–99
Deorah S, Lynch CF, Sibenaller ZA, Ryken TC (2006) Trends in brain cancer incidence, survival in the United States: Surveillance, Epidemiology, End Results Program, 1973 to 2001. Neurosurg Focus 20:E1
El Andaloussi A, Lesniak MS (2006) An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol 8:234–243
El Andaloussi A, Lesniak MS (2007) CD4+CD25+FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J Neurooncol 83:145–152
El Andaloussi A, Han Y, Lesniak MS (2006) Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J Neurosurg 105:430–437
Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE 2nd, Bigner DD, Dranoff G, Sampson JH (2006) Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 66:3294–3302
Garcia-Lora A, Algarra I, Garrido F (2003) MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol 195:346–355
Golgher D, Jones E, Powrie F, Elliott T, Gallimore A (2002) Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur J Immunol 32:3267–3275
Guevara P, Sotelo J (1999) C6 rat glioma grown into the peritoneal cavity, a large source of tumoral cells for subcutaneous transplant of glioma. J Neurooncol 44:91–92
Hart MN, Linthicum DS, Waldschmidt MM, Tassell SK, Schelper RL, Robinson RA (1987) Experimental autoimmune inflammatory myopathy. J Neuropathol Exp Neurol 46:511–521
He J, Gurunathan S, Iwasaki A, Ash-Shaheed B, Kelsall BL (2000) Primary role for Gi protein signaling in the regulation of interleukin 12 production and the induction of T helper cell type 1 responses. J Exp Med 191:1605–1610
Hofstetter HH, Shive CL, Forsthuber TG (2002) Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund’s adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol 169:117–125
Hou W, Wu Y, Sun S, Shi M, Sun Y, Yang C, Pei G, Gu Y, Zhong C, Sun B (2003) Pertussis toxin enhances Th1 responses by stimulation of dendritic cells. J Immunol 170:1728–1736
Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H (2003) Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res 9:4404–4408
Jager D, Jager E, Knuth A (2001) Immune responses to tumour antigens: implications for antigen specific immunotherapy of cancer. J Clin Pathol 54:669–674
Kohno S, Munoz JA, Williams TM, Teuscher C, Bernard CC, Tung KS (1983) Immunopathology of murine experimental allergic orchitis. J Immunol 130:2675–2682
Li M, Liu J, Zhang SZ, Zhou Y, Guo YW, Chen Q, Ke YQ, Jiang XD, Cai YQ (2011) Cellular immunologic response to primary cryoablation of C6 gliomas in rats. Technol Cancer Res Treat 10:95–100
Linehan DC, Goedegebuure PS (2005) CD25+CD4+ regulatory T-cells in cancer. Immunol Res 32:155–168
Linthicum DS (1982) Development of acute autoimmune encephalomyelitis in mice: factors regulating the effector phase of the disease. Immunobiology 162:211–220
Linthicum DS, Frelinger JA (1982) Acute autoimmune encephalomyelitis in mice. II. Susceptibility is controlled by the combination of H-2 and histamine sensitization genes. J Exp Med 156:31–40
Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC (2002) Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 169:2756–2761
Lopez-Gonzalez MA, Sotelo J (2000) Brain tumors in Mexico: characteristics and prognosis of glioblastoma. Surg Neurol 53:157–162
Mascart F, Verscheure V, Malfroot A, Hainaut M, Pierard D, Temerman S, Peltier A, Debrie AS, Levy J, Del Giudice G, Locht C (2003) Bordetella pertussis infection in 2-month-old infants promotes type 1 T cell responses. J Immunol 170:1504–1509
Mielcarek N, Hornquist EH, Johansson BR, Locht C, Abraham SN, Holmgren J (2001) Interaction of Bordetella pertussis with mast cells, modulation of cytokine secretion by pertussis toxin. Cell Microbiol 3:181–188
Munoz JJ, Bernard CC, Mackay IR (1984) Elicitation of experimental allergic encephalomyelitis (EAE) in mice with the aid of pertussigen. Cell Immunol 83:92–100
Nencioni L, Pizza MG, Volpini G, De Magistris MT, Giovannoni F, Rappuoli R (1991) Properties of the B oligomer of pertussis toxin. Infect Immun 59:4732–4734
Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E (1999) Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 59:3128–3133
Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM, Greaves DR (2008) Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol 28:433–440
Pineda B, Estrada-Parra S, Pedraza-Medina B, Rodriguez-Ropon A, Perez R, Arrieta O (2005) Interstitial transfer factor as adjuvant immunotherapy for experimental glioma. J Exp Clin Cancer Res 24:575–583
Ryan M, McCarthy L, Rappuoli R, Mahon BP, Mills KH (1998) Pertussis toxin potentiates Th1 and Th2 responses to co-injected antigen: adjuvant action is associated with enhanced regulatory cytokine production and expression of the co-stimulatory molecules B7–1, B7–2 and CD28. Int Immunol 10:651–662
Shimizu J, Yamazaki S, Sakaguchi S (1999) Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol 163:5211–5218
Shive CL, Hofstetter H, Arredondo L, Shaw C, Forsthuber TG (2000) The enhanced antigen-specific production of cytokines induced by pertussis toxin is due to clonal expansion of T cells and not to altered effector functions of long-term memory cells. Eur J Immunol 30:2422–2431
Strojnik T, Kavalar R, Zajc I, Diamandis EP, Oikonomopoulou K, Lah TT (2009) Prognostic impact of CD68 and kallikrein 6 in human glioma. Anticancer Res 29:3269–3279
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Tomayko MM, Reynolds CP (1989) Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 24:148–154
Tonon S, Goriely S, Aksoy E, Pradier O, Del Giudice G, Trannoy E, Willems F, Goldman M, De Wit D (2002) Bordetella pertussis toxin induces the release of inflammatory cytokines and dendritic cell activation in whole blood: impaired responses in human newborns. Eur J Immunol 32:3118–3125
Velasquez-Perez L, Jimenez-Marcial ME (2003) Clinical-histopathologic concordance of tumors of the nervous system at the Manuel Velasco Suarez National Institute of Neurology and Neurosurgery in Mexico City. Arch Pathol Lab Med 127:187–192
Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH (2001) Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61:4766–4772
Zhu X, Lu C, Xiao B, Qiao J, Sun Y (2005) An experimental study of dendritic cells mediated immunotherapy against intracranial gliomas in rats. J Neurooncol 74:9–17
Acknowledgments
This work was partially supported by CONACyT-México grants (90550) and ICyTDF (PICSA10-143).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Orozco-Morales, M., Sánchez-García, FJ., Guevara-Salazar, P. et al. Adjuvant immunotherapy of C6 glioma in rats with pertussis toxin. J Cancer Res Clin Oncol 138, 23–33 (2012). https://doi.org/10.1007/s00432-011-1069-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-011-1069-y