
Abstract
The precise role of different toll-like receptor (TLR) superfamily members is just beginning to get elucidated in glioblastoma multiforme (GBM). In this study, we observed heightened TLR4 levels in GBM tumor samples as compared to adjacent normal tissue. Since the pro-inflammatory cytokine tumor necrosis factor (TNF)α induces NF-κB activation in GBM, and as several common signaling mediators are involved in TNFα and TLR4-mediated NF-κB activation, we investigated the role of TLR4 in the regulation of NF-κB activation and inflammatory responses in TNFα-treated glioma cells. TNFα elevated TLR4 expression and inhibition of TLR4 signaling by either signaling inhibitor, neutralizing antibody, or small interfering RNA (siRNA)-attenuated TNFα-induced NF-κB activation. TLR4-mediated NF-κB activation was independent of canonical myeloid differentiation factor 88 signaling but involved toll/IL-1R homology domain-containing adaptor protein-inducing interferon-β. Inhibition of TLR4 signaling abrogated TNFα-induced increase in (1) transcription factors interferon (IFN) regulatory factor 3 and STAT-1 and (2) IFNβ and inflammatory cytokines/chemokines expression. Furthermore, TNFα-induced TLR4-dependent increase in AKT activation and HIF-1α transcriptional activation suggested the existence of TLR4–AKT–HIF-1α axis. Importantly, TNFα-induced TLR4 was abrogated in cells transfected with dominant negative IκB and HIF-1α siRNA. Our studies indicate that TNFα triggered TLR4–HIF-1α and NF-κB–TLR4 feed-forward loops act in tandem to sustain inflammatory response in glioma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although members of the toll-like receptor (TLR) superfamily are associated with pathogen recognition and activation of innate immunity, recent studies have indicated their involvement in tumor progression and chemoresistance [1]. The importance of TLR4 in ovarian [2, 3], head and neck [4], and lung cancer [5] is well established. These studies have clearly indicated a link between TLR4 signaling, inflammation, chemoresistance, and sensitivity to apoptosis in cancer [2–4]. Although TLR4 signals through myeloid differentiation factor 88 (MyD88) leads to activation of NF-κB [6], it can also induce MyD88-independent signaling that activates late phase of NF-κB as well as interferon (IFN) regulatory factor 3 (IRF3) which triggers the production of IFNβ [6]—a regulator of inflammatory response [7]. TLR4 activation also induces the production of pro-inflammatory cytokines including tumor necrosis factor (TNF)α [8].
Constitutive activation of NF-κB in glioblastoma multiforme (GBM) is associated with enhanced expression of genes that facilitate tumorigenesis [9]. Constitutively active NF-κB triggers TNFα-dependent inflammation [10]. We have recently reported that NF-κB is a direct modulator of HIF-1α [11], a transcription factor involved in the regulation of several genes implicated in angiogenesis, metabolism, and cell survival [12]. Because of its critical involvement in cancer progression the NF-κB signaling pathway has become a potential target for pharmacological intervention [13]. Given its central role in cancer biology, HIF-1α is considered to be another most important molecular target in the treatment of cancer [14]. Importantly, inflammation is an important contributing factor in cancer development [15] and HIF-1α is a critical link between inflammation and tumorigenesis [11, 16]. Besides, HIF-1α is overexpressed in GBM [17, 18].
Chronic inflammation is a potential risk factor for tumor progression and activation of TLR induces inflammatory factors [19, 20]. Lipopolysaccharide (LPS)-induced TLR4-mediated HIF-1α regulates release of pro-inflammatory cytokines [21]. Also, various signaling mediators/adaptors that participate in tumor necrosis factor receptor (TNFR) signaling leading to NF-κB activation are also involved in TLR4 signaling [22] and TNFα antagonist Remicade attenuates TLR4-mediated inflammatory responses [8].
Given that (1) several common signaling mediators are involved in TNF receptor and TLR4-mediated NF-κB activation, (2) the sheer complexity of signaling events that activates NF-κB in addition to canonical TNF receptor signaling, (3) NF-κB is a modulator of HIF-1α expression in GBM, (4) both inflammation and HIF-1α are indispensable components in GBM progression, and (5) TNFα promotes a pro-inflammatory milieu in glioma cells which are resistant to TNFα-induced apoptosis, we explored the importance of TLR4 in regulating signaling events that promote inflammatory responses in TNFα-treated glioma cells.
To our knowledge, this is the first report profiling the expression of TLR4 in GBM tumors. Our studies indicate that MyD88-independent but adaptor protein-inducing interferon-β (TRIF)-dependent TLR4 signaling is a critical component of TNFα-induced NF-κB activation. The primary finding of our study is the uncovering of a critical role of NF-κB in TLR4 activation. This induction of TLR4 by NF-κB coupled with the ability of TLR4 to regulate NF-κB highlights the existence of NF-κB–TLR4 loop. We also demonstrate the existence of AKT driven HIF-1α–TLR4 feed-forward loop in TNFα-treated glioma cells. These signaling pathways triggered by TLR4 upon TNFα stimulation act in concert to sustain and amplify inflammatory response in glioma cells.
Materials and methods
Processing of tissue and immunohistochemistry
Tissue samples were collected from patients with histologically confirmed GBM (n = 19) as per the guidelines of Institutional Human Ethics Committee of National Brain Research Centre. During surgery, non-neoplastic brain tissue (n = 7) was obtained from margins of the tumors whenever possible and was used as control. Tumor and nontumor tissues were washed in phosphate-buffered saline (PBS), fixed with 10% formalin for 24–30 h and left in 30% sucrose for 24–30 h. The cryotome sections (20 μM thick) were taken on gelatin-coated slides and processed for immunohistochemistry.
Tissues sections were washed thrice with PBS, quenched using 3% H2O2 for 10 min at 25°C, washed and blocked in 2% bovine serum albumin, 3% normal goat serum, and 0.1% TX-100 for 1 h at 25°C. After blocking, sections were incubated with anti-TLR4 antibody in blocking buffer overnight at 4°C. After primary antibody incubation, sections were washed with PBS containing 0.01% Tween-20 and incubated with biotinylated secondary antibody in PBS for 2 h at 25°C. Sections were then washed and incubated for 1.5 h at 25°C with avidin–biotin complex (Vector) as per kit instructions. After washes, sections were developed with Nova Red substrate (Vector) for 3–5 min, rinsed with double distilled water, washed and counter-stained with Mayer’s hematoxylin for 10–15 s. Sections were then rinsed with tap water and washed with double distilled water, dried overnight, mounted with DPX mounting media and observed through Leica DMRXA2 microscope.
Cell culture and treatment
Glioblastoma cell lines A172 and LN229 obtained from the American Type Culture Collection were cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum. On attaining semiconfluence, cells were switched to serum-free media (SFM) and after 6 h, cells were treated with 50 ng/ml TNFα (R&D) in SFM in the presence or absence of either TLR neutralizing antibody (Imgenex), or TRIF inhibitory peptide (InVivogen) or TLR4 signaling inhibitor TAK-242 (InVivogen) or AKT inhibitor LY294002. All reagents were purchased from Sigma unless otherwise stated. The dominant negative (DN) MyD88 and TNF receptor-associated factor 6 (TRAF6) constructs were a kind gift from Luke A.J. O’Neill (Trinity College, Ireland) [23]. The HIF-1α luciferase reporter was gifted by Chinmay Mukhopadhyay (JNU, India) [24]. NF-κB reporter and DN-IκB construct was purchased from Clontech.
Western blot analysis
Whole cell lysates and nuclear extracts were isolated from cells treated with or without TNFα in the presence and absence of TLR4 neutralizing antibody, TLR4 signaling inhibitor TAK-242, or TRIF inhibitory peptide or LY294002 as described previously [11]. Small amount of tumor tissues were homogenized in the same whole cell lysate buffer used for Western blot analysis and samples were stored in −20°C until further use. Lysates were electrophoresed on 7–12% polyacrylamide gel and Western blot analysis was performed as described [11] using the following antibodies—TLR4, MyD88 (Anaspec), IRAK4 (Anaspec), TIRAP, TRAF6, TRIF, IRF3, pSTAT1, STAT1, AKT, pAKT, and HIF-1α (BD biosciences). Antibodies to C23 and peroxidase conjugated β-actin were purchased from Santa Cruz and Sigma, respectively. Antibodies were purchased from Cell Signaling unless otherwise mentioned.
Detection of TLR4 expression by fluorescence-activated cell sorting (FACS)
TLR4 expression on glioma cells treated with TNFα for 24 h was measured in an immunofluorescence flow cytometry assay as described previously [25]. Briefly, 106 cells were incubated with anti-TLR4 for 40 min at 4°C. Following incubation, cells were washed and incubated with secondary Ab (anti-rabbit conjugated to FITC). After a further incubation for 30 min on ice, cells were washed, resuspended in PBS and staining was measured by flow cytometry on FACS Calibur (BD Biosciences) using the CellQuestPro analysis software (Becton Dickinson, Mountain View, CA, USA).
Luciferase assay
Cells at ∼60–70% confluence in 24-well plates were transiently transfected with 0.3 μg NF-κB or HIF-1α reporter plasmids and 10 ng of the Renilla luciferase expression vector pRL-TK as a transfection control using Lipofectamine 2,000 (Life Technologies-Invitrogen) as described previously [11]. Following transfection for 18–24 h, cells were serum starved for 4 h followed by treatment with 50 ng/ml TNFα for 6–24 h in the presence and absence of either 1 μg/ml TLR4 neutralizing antibody, or 5 μM TLR4 signaling inhibitor TAK-242, or 20 μM TRIF control/inhibitory peptide or 10 μM AKT inhibitor. Luciferase activity was measured using Dual luciferase assay kit according to the manufacturer’s protocol (Promega) with GloMax 96 microplate luminometer. The results are expressed as fold change in activity over control. For determining NF-κB transcriptional activity in presence of dominant negative MyD88 and TRAF6 constructs, cells were co-transfected with 0.3 μg of either DN–MyD88 or DN–TRAF6 constructs along with NF-κB luciferase reporter constructs. Similarly, HIF-1α luciferase activity was determined in cells co-transfected with DN–IκB construct or treated with AKT inhibitor. Co-transfection experiments with DN constructs were compared with control transfection using the appropriate empty vectors for each construct.
Small interfering RNA transfection
Small interfering RNA (siRNA)-mediated knock down of TLR4 or HIF-1α was performed as described previously [11]. Briefly, 18 h prior to transfection 3 × 104 cells were seeded onto 24-well plates in medium without antibiotics and transfection of siRNAs was carried out with Lipofectamine RNAiMax (Life Technologies–Invitrogen). All transfections were carried out with 50 nmol/L duplex siRNA. The siRNA duplexes were purchased from Thermo Fischer Scientific. Nonspecific siRNA that does not target any known mammalian gene was purchased from Proligo (Singapore). For luciferase reporter assay of TLR4 siRNA-transfected cells, these cells were again transfected with NF-κB reporter and Renilla luciferase constructs 18 h post-siRNA transfection and were subsequently processed for NF-κB reporter assay. Similarly, cells were harvested 24 h following transfection with HIF-1α siRNA and Western blotting performed to determine TLR4 levels.
IFNβ ELISA
Human IFNβ enzyme-linked immunosorbent assay (ELISA) kit (Pierce) was used to determine the concentration of IFNβ in supernatant collected from cells treated with TNFα in the presence or absence of TLR4 signaling inhibitor according to the manufacturer’s instructions.
Cytokine and chemokine bead array
Cytokine and chemokine bead array kit (Human Inflammation CBA kit; BD Biosciences, NJ, USA) was used to quantitatively measure cytokine and chemokine levels in the supernatant collected from cells treated with TNFα in the presence or absence of TLR4 signaling inhibitor as described [26].
Statistical analysis
One-way analysis of variance was used to identify statistical differences between groups. For these purposes, SigmaStat software version 3.5 (Systat Software Inc., San Jose, CA) was used. A value of p < 0.05 was considered significant.
Results
Elevated TLR4 levels in glioblastoma biopsy samples
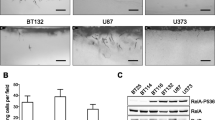
To demonstrate the role of TLR4 signaling in GBM, we first examined the expression of TLR4 in GBM tumor samples. Immunohistochemistry revealed TLR4 immunolocalization in GBM tumors (Fig. 1a). Western blot analysis demonstrated elevated TLR4 levels in the lysates of GBM tumor tissues, as compared to the adjacent non-neoplastic tissue (Fig. 1b).

TNFα induces TLR4 expression in glioma cells. a TLR4 immunolocalization in glioma tumor samples. Cryosections of glioma and adjacent normal tissues were immunostained for TLR4 as described in “Materials and methods” section. Images taken at ×40 magnification. Inset TLR4 immunolocalization at ×100. b Western blot analysis demonstrating elevated TLR4 expression in GBM tumor as compared to surrounding non-neoplastic tissue. c TNFα increases TLR4 expression in glioma cells as demonstrated by Western blot. The figure is a representative blot from three independent experiments with identical results. Blots were reprobed with β-actin to establish equivalent loading. Densitometric measurements were performed on individual immunoblots and values indicate protein level normalized to its corresponding β-actin level. The values represent the means ± SEM from three individual experiments. Asterisk significant change from control (p < 0.05). d FACS analysis indicates elevated TLR4 levels in glioma cells upon treatment with TNFα for 24 h. A representative histogram is shown from two independent experiments with identical trend
TNFα induces TLR4 expression in glioma cells
NF-κB activation is involved in LPS-induced TLR4 expression [27]. Since NF-κB is constitutively expressed in GBM [9] and as TNFα induces NF-κB activation in glioma cells [25], we investigated the status of TLR4 in TNFα-treated glioma cells. Western blot analysis indicated an increase in TLR4 expression upon TNFα treatment in a time-dependent manner (Fig. 1c). FACS analysis also indicated an increase in TLR4 expression in TNFα-treated glioma cells. The expression of TLR4 on the surface of untreated glioma cells (black line) were elevated in the presence of TNFα (gray line; Fig. 1d).
TLR4 is involved in TNFα-mediated NF-κB activation in glioma
We have previously reported that TNFα induces NF-κB transcriptional activity in glioma cells [25]. Transfection of glioma cells with NF-κB reporter construct followed by TNFα treatment for 3, 6, 12, and 24 h resulted in ∼6-, 9-, 7-, and 5-fold increase in NF-κB activity in A172 glioma cells (Fig. 2a). Similar trend was observed in LN229 (Fig. 2a). Although elevated NF-κB activity persisted at later time intervals, maximal NF-κB activity was observed at 6 h post-TNFα treatment (Fig. 2a). We therefore measured NF-κB activity at 6 h for subsequent experiments. As TLR4 signaling activates NF-κB [28], we determined the involvement of TLR4 in TNFα-induced NF-κB activation by three independent strategies. These involved treatment of glioma cells with either TLR4 downstream signaling inhibitor TAK-242 [29] or TLR4 neutralizing antibody or TLR4 siRNA. TNFα-mediated increase in NF-κB activity was decreased by ∼40–50% in cells treated with TAK-242 (Fig. 2b). Neutralization of TLR4 using anti-TLR4 neutralizing antibody resulted in a significant 30% decrease in TNFα-induced NF-κB activity (Fig. 2c). siRNA-mediated knockdown of TLR4 decreased TNFα-mediated increase in NF-κB expression as compared to cells transfected with nonspecific siRNA (Fig. 2d). Decreased TLR4 levels upon transfection with TLR4 siRNA is shown in Fig. 2d inset. These results suggest that TLR4-mediated signaling contributes to TNFα-induced NF-κB activation in glioma cells. However, the inability of TAK-242, TLR4 neutralizing antibody, and TLR4 siRNA to completely abrogate TNFα-induced NF-κB activation indicates that other TNFR-mediated pathways independent of TLR4 are involved in TNFα-induced NF-κB activation.

TNFα induces TLR4-NF-κB feed-forward loop in glioma cells. a TNFα increases NF-κB transcriptional activity in glioma cells. Following transfection of glioma cells with NF-κB reporter constructs, cells were treated with TNFα for different time intervals and NF-κB activity was determined. The graph represents fold change in activity over control. b–d TNFα-induced NF-κB transcriptional activity in glioma cells is abrogated upon inhibition of TLR4 signaling. Following transfection of glioma cells with NF-κB reporter construct, cells were treated with TNFα in the presence and absence of b TLR4 signaling inhibitor TAK-242 or c TLR4 neutralizing antibody or d TLR4 siRNA for 6 h and reporter assay was performed to determine NF-κB activity. The graph represents fold change in activity over control. Values in a–d represent the means ± SEM from three independent experiments. Asterisk significant increase from untreated control, number sign significant decrease from TNFα-treated cells (p < 0.05). The inset in 2 d shows TLR4 levels in cells transfected with TLR4 siRNA and nonspecific (NS) siRNA as analyzed by Western blot. e TNFα-induced NF-κB regulates TLR4 expression. Western blot demonstrates decreased TLR4 expression in TNFα-treated glioma cells transfected with DN-IκB as compared to mock-transfected control. The figure is representative of three independent experiments. Blots were reprobed for β-actin to establish equivalent loading. Asterisk significant increase from untreated control, number sign significant decrease from TNFα-treated cells (p < 0.05)
Existence of NF-κB–TLR4 feed-forward loop in glioma cells
Upon stimulation with TNF-alpha, a positive signaling feedback loop in the NF-κB pathway prolongs LPS-induced gene expression [28]. Interestingly, disruption of NF-κB signaling attenuates LPS-mediated TLR4-dependent innate immune responses upon infection with opportunistic pathogen [30]. We therefore investigated whether TNFα-induced NF-κB triggers positive signaling in the TLR4–NF-κB pathway. TNFα-induced TLR4 expression was abrogated in cells transfected with DN–IκB (Fig. 2e). This, together with the ability of TLR4 signaling inhibitor to abrogate NF-κB activity, suggests the existence of a TLR4–NF-κB feed-forward loop in TNFα-treated glioma cell.
TNFα increases expression of molecules associated with MyD88-dependent TLR4–NF-κB signaling
Activation of TLR4 leads to stimulation of MyD88-dependent and MyD88-independent pathways. In the canonical MyD88-dependent pathway, association of MyD88 to TLR4 results in the recruitment of interleukin-1 receptor-associated kinase (IRAK)1, IRAK4, TRAF6, which subsequently results in the activation of NF-κB [6]. The interaction of MyD88 adapter-like (Mal)/TIRAP with TRAF6 is critical for NF-κB activation mediated by TLR4 signaling [31]. Since TLR4 was elevated upon TNFα treatment and as TNFα-induced NF-κB activity involves TLR4, we determined the expression of accessory molecules that are required for the formation of protein complex crucial to NF-κB activation downstream of MyD88–TLR4 signaling. Treatment with TNFα elevated the expression of MyD88, TIRAP, and TRAF6 (Fig. 3a).

TLR4 regulates NF-κB activation in a MyD88-independent manner. a Expression of molecules associated with MyD88-dependent TLR4–NF-κB signaling in TNFα-treated A172 glioma cells. The expressions of MyD88, TIRAP, IRAK4 and TRAF6 in A172 cells treated with TNFα for different intervals of time were analyzed by Western blotting. A representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading. b TLR4-induced NF-κB activation is MyD88 independent. Glioma cells co-transfected with DN–MyD88 and NF-κB reporter constructs were treated with TNFα for 6 h and luciferase reporter assay was performed to determine NF-κB activity. The graph represents fold change in activity over control. c Transfection with DN–TRAF6 abrogates TNFα-induced NF-κB activation. Glioma cells co-transfected with DN–TRAF6 and NF-κB reporter constructs were treated with TNFα for 6 h and luciferase reporter assay was performed to determine NF-κB activity. The graph represents fold change in activity over control. Values in b and c represent the means ± SEM from three independent experiments. Asterisk significant increase from untreated control, number sign significant decrease from TNFα-treated cells (p < 0.05)
TNFα induces NF-κB activation in a MyD88-independent manner
Since TNFα increased expression of different components of MyD88-dependent TLR4 signaling that activates NF-κB activation, we questioned whether NF-κB activation is MyD88 dependent. To address this question, we determined NF-κB transcriptional activity in cells transfected with DN–MyD88 in the presence and absence of TNFα. Transfection of cells with DN–MyD88 had no effect on TNFα-mediated increase in NF-κB activation (Fig. 3b) indicating that TLR4 driven NF-κB activation is independent on MyD88.
As TRAF6 mediates both early and late phase of NF-κB activation by cooperating with MyD88 and TRIF, respectively [32], its role in TNFα-induced NF-κB activation was determined in cells transfected with DN–TRAF6 in the presence and absence of TNFα. A decrease in TNFα-induced NF-κB activity was observed in cells transfected with DN–TRAF6 (Fig. 3c). This involvement of TRAF6 indicated the existence of MyD88-independent and TRIF-NF-κB pathway, respectively [32] in TLR4-mediated NF-κB activation.
Involvement of TRIF in TNFα-induced NF-κB activation
Since TLR4 induces NF-κB activation through adaptor molecule TRIF independent of MyD88, we next determined the importance of TRIF in NF-κB activation. An increase in TRIF expression was observed in TNFα-treated glioma cells (Fig. 4a). To investigate the role of TRIF on TNFα-induced NF-κB activation, we determined NF-κB activity in cells treated with TRIF inhibitory or control peptide in the presence and absence of TNFα While treatment with control TRIF peptide had no effect on TNFα-induced NF-κB activation, a 30% decrease in TNFα-induced NF-κB transcriptional activity was seen in both A172 and LN229 cells in the presence of TRIF inhibitory peptide (Fig. 4b).

Involvement of TRIF in TLR4-mediated NF-κB activation. a TNFα increases the expression of TRIF in glioma cells. Western blot demonstrates increase in TRIF expression in TNFα-treated glioma cells. A representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading. Densitometric measurements were performed on individual immunoblots and values indicate protein level normalized to its corresponding β-actin level. b TNFα-induced NF-κB transcriptional activity in glioma cells, is abrogated upon treatment with TRIF inhibitory peptide. Following transfection of glioma cells with NF-κB reporter construct, cells were treated with TNFα in the presence and absence of TRIF inhibitory or control peptide for 6 h and reporter assay was performed to determine NF-κB activity. The graph represents fold change in activity over control. Values represent the means ± SEM from three independent experiments. Asterisk significant increase from untreated control, number sign significant decrease from TNFα-treated cells (p < 0.05)
TNFα induces IRF3 in a TLR4-dependent manner
MyD88-independent signaling can activate late phase of NF-κB as well as IRF3 that subsequently triggers IFNβ [6]. Since MyD88-independent TRIF signaling is involved in TNFα-mediated NF-κB activation, we determined the status of IRF3 in TNFα-treated cells. Elevated nuclear IRF3 level was observed in glioma cells upon increasing exposure to TNFα (Fig. 5a). This increase in IRF3 expression is dependent on TLR4, since TAK-242 decreased TNFα-mediated increase in IRF3 expression (Fig. 5b).

TLR4 regulate TNFα-mediated increase in IRF3, STAT1 and IFNβ expression. a Increased IRF3 expression in glioma cells upon TNFα treatment. Western blot demonstrates an increase in nuclear IRF3 expression in glioma cells treated with TNFα for 24 h. b TNFα-induced increase in IRF3 is dependent on TLR4. Glioma cells were treated with TNFα in the presence and absence of TAK-242 for 24 h and Western blot analysis was performed on nuclear extracts to determine IRF3 expression. a, b Representative blots are shown from three independent experiments with identical results. Blots were reprobed for C23 to establish equivalent loading. Densitometric measurements were performed on individual immunoblots and values indicate protein level normalized to its corresponding C23 level. c Inhibition of TLR4 signaling reduces TNFα-induced increase in IFNβ release. The graph shows IFNβ levels in picogram per millilter in TNFα-treated glioma cells in the presence and absence of TAK-242 as analyzed by ELISA. Values represent mean ± SEM from three individual experiments. Asterisk significant increase from control, number sign significant decrease from TNFα-treated cells (p < 0.05). d Western blot analysis reveals a time dependent increase in STAT1 Ser phosphorylation in TNFα-treated glioma cells. Densitometric measurements were performed on individual immunoblots and values indicate protein level normalized to its corresponding STAT1 and β-actin levels. e TNFα-induced STAT1 phosphorylation is TLR4 dependent. Cells were treated with TNFα in the presence and absence of TLR4 signaling inhibitor for 24 h and Western blot was performed to determine the expression of pSTAT1. The figure is representative of three independent experiments. Blots were reprobed for β-actin to establish equivalent loading
TLR4 regulates increased IFNβ expression in glioma cells
As TLR4-induced MyD88-independent signaling lead to IFNβ production [33], we determined IFNβ level in TNFα-treated cells. ELISA indicated an increase in IFNβ levels upon TNFα treatment in a TLR4-dependent manner (Fig. 5c), as TLR4 signaling inhibitor TAK-242 decreased TNFα-mediated increase in IFNβ levels (Fig. 5c).
TNFα induces TLR4–STAT1 axis in glioma cells
As TNFα-induced IFNβ–STAT1 pathway is crucial for sustaining and amplifying the expression of TNFα-induced genes which promote a feed-forward cycle of inflammatory response [34] and since TLR4 induces Ser-727 STAT1 phosphorylation [35], we investigated pSTAT1 expression in TNFα-treated glioma cells. An increase in pSTAT1 was observed upon increasing exposure to TNFα (Fig. 5d). This increase in pSTAT1 was TLR4 dependent, as inhibition of TLR4 signaling resulted in decrease in TNFα-induced increase in pSTAT1 levels (Fig. 5e). We next determined whether TNFα-induced increase in pSTAT1 is NF-κB dependent. Transfection with DN–IκB has no effect on TNFα-induced STAT1 expression (data not shown). We also investigated whether TLR4 mediates its own expression through STAT1, by determining its level in cells treated with TNFα in the presence and absence of STAT1 inhibitor Fludarabine. Western blot analysis indicated that TNFα-induced TLR4 expression is independent of STAT1 activation (data not shown).
TNFα induces TLR4–HIF-1α feed-forward loop in glioma cells
HIF-1α regulates TLR expression [36] and HIF-1α activation upon LPS stimulation is TLR4 dependent [21]. Besides, NF-κB regulates HIF-1α transcriptional activation [37] and we have recently reported that HIF-1α activation in glioma cells is NF-κB dependent [11]. We therefore investigated the involvement of TLR4 in HIF-1α induction and vice versa in TNFα-treated cells. TNFα elevated HIF-1α transcriptional activity in glioma cells (Fig. 6a). Interestingly, inhibition of TLR4 signaling abrogated TNFα-induced HIF-1α activation (Fig. 6a). To determine whether the reverse was true, TLR4 level was determined in cells transfected with HIF-1α siRNA. Transfection with HIF-1α siRNA prevented TNFα-mediated induction of TLR4 expression in glioma cells (Fig. 6b). Abrogation of HIF-1α activity upon inhibition of TLR4 signaling along with the ability of HIF-1α to regulate TNFα-induced TLR4 expression suggests the existence of HIF-1α-TLR4 feed-forward loop in TNFα-treated glioma cells.

Existence of TLR4–AKT–HIF-1α axis in TNFα-treated glioma cells. a Inhibition of TLR4 signaling abrogates TNFα-induced HIF-1α transcriptional activity in TNFα-treated cells. Following transfection of glioma cells with HIF-1α reporter construct, cells were treated with TNFα in the presence and absence of TAK-242 for 24 h and reporter assay was performed to determine HIF-1α activity. The graph represents fold change in activity over control. b siRNA-mediated knockdown of HIF-1α prevents TNFα-mediated increase in TLR4 expression. Cells transfected with HIF-1α siRNA or nonspecific siRNA were treated with TNFα for 24 h and Western blot analysis was performed to determine TLR4 expression. Inset Western blot indicates HIF-1α expression in cells transfected with HIF-1α and nonspecific (NS) siRNA, to indicate specificity of the HIF-1α siRNA. c TNFα-induced increase in AKT phosphorylation is abrogated upon inhibition of TLR4 signaling. Glioma cells were treated with TNFα in the presence of TAK-242 for 24 h and Western blot was performed to determine the levels of pAKT. Representative blot is shown from three independent experiments with identical results. Blots were reprobed for β-actin to establish equivalent loading. d Treatment with AKT inhibitor abrogates HIF-1α transcriptional activity in TNFα-treated cells. Following transfection with HIF-1α reporter construct, cells were treated with TNFα in the presence and absence of 10 μM of AKT inhibitor LY294002 for 24 h and reporter assay was performed to determine HIF-1α activity. The graph represents fold change in activity over control. b and c Densitometric measurements were performed on individual immunoblots for each antibody tested and values indicate protein level normalized to its corresponding β-actin and AKT levels. Values in a and d represent the means ± SEM from three independent experiments. Asterisk Significant increase from untreated control, number sign significant decrease from TNFα-treated cells (p < 0.05)
Existence of TLR4–AKT–HIF-1α axis in TNFα-treated glioma cells
AKT is phosphorylated upon TLR4 signaling [38] and AKT regulates HIF-1α activity [39]. As we have reported that TNFα increases AKT phosphorylation in glioma cells [40], we investigated whether TNFα-induced TLR4 is involved in AKT phosphorylation. We determined AKT levels in TNFα-treated cells in the presence and absence of TAK-242. Inhibition of TLR4 signaling abrogated TNFα-induced increase in AKT phosphorylation (Fig. 6c). Importantly, TNFα-induced AKT regulates HIF-1α transcriptional activity, as HIF-1α activity was abrogated in cells treated with AKT inhibitor (Fig. 6d). This indicates that a TLR4–AKT–HIF-1α axis exists in glioma cells alongside the TLR4–NF-κB feed-forward loop.
TLR4 promotes inflammatory milieu in TNFα-treated glioma cells
TNFα-induced NF-κB activation is crucial for tumor progression as it induces the expression of the pro-inflammatory cytokines [41]. Since TAK-242 selectively inhibits TLR4-mediated cytokine production [29], we performed cytometric bead array to determine the expression of inflammatory cytokines released from cells treated with TNFα in the presence and absence of TAK-242. Treatment with TNFα increased expression of cytokines IL-1β, IL-6, IL-8, IL-10, and IL-12 in glioma cells (Fig. 7a). Though inhibition of TLR4 signaling had no effect on TNFα-induced IL-1β expression, levels of IL-6, IL-8, IL-10, and IL-12 was abrogated in the presence of TAK-242 (Fig. 7a). Thus, TNFα-induced TLR4 regulates the release of both immunostimulatory and immunosuppressive cytokines in glioma cells. Similarly, TNFα increased expression of chemokines MCP-1, MIG-1, and RANTES in glioma cells in TLR4-dependent manner (Fig. 7b). However, TNFα-induced increase in chemokine inducible protein-10 (IP-10) upon was not affected in the presence of TAK-242 (Fig. 7b). Taken together, these results indicate that different signaling circuitries triggered by TLR4 upon TNFα stimulation work in tandem to sustain an inflammatory milieu in glioma cells (Fig. 7c).

TLR4 regulates pro-inflammatory response in glioma cells. Increase in inflammatory cytokine/chemokine observed upon TNFα treatment was significantly suppressed upon inhibition of TLR4 signaling. Expression of inflammatory cytokines a and chemokines b in A172 and LN229 cells treated with different combination of TNFα and TAK-242 for 24 h, as observed by cytometric bead array. Values a and b represent mean ± SEM from three individual experiments. Asterisk significant increase from control, number sign significant decrease from TNFα-treated cells (p < 0.05). c Proposed model indicating that TLR4 activation induces NF-κB through a MyD88-independent and TRIF-dependent pathway. In addition, TNFα induces a TLR4–STAT1 and TLR4–AKT–HIF-1α axis. Importantly, TNFα triggers the NF-κB–TLR4 and TLR4–HIF-1α feed-forward loops that operate concurrently to sustain TLR4-mediated release of inflammatory mediators
Discussion
TLR4 signaling culminates in the activation of NF-κB that promotes a pro-inflammatory and pro-growth microenvironment [1, 42]. We have reported that TNFα triggers the assembly of canonical TNFR-dependent signaling complex that participates in TNFα-mediated NF-κB activation in glioma cells [25]. Importantly, TNFα- and TLR4-mediated signaling events leading to NF-κB activation share several common mediators. As the link between TLR4 signaling and inflammation in cancer is documented [2–4], we explored the role of TLR4 in the regulation of inflammatory response in GBM. We not only report for the first time a heightened TLR4 levels in GBM tumors as compared to normal brain tissue but also demonstrate that TNF-α increases TLR4 expression in glioma cells.
In addition to TLR4-activated conserved MyD88-dependent pathway that lead to NF-κB activation, alternative MyD88-independent pathways involving different adaptor molecules TRIF/TRAM also trigger TLR4-mediated NF-κB activation [42]. Inhibition of TLR4 signaling abrogated TNFα-induced NF-κB activation in glioma cells. Despite increased expression of the components associated with MyD88-dependent TLR4-NF-κB signaling, TLR4-induced NF-κB activation in a MyD88-independent manner. The subtle increase in TLR4 expression in A172 cells upon TNFα treatment could have resulted from high basal level of TLR4 in these glioma cells due to elevated constitutive NF-κB expression. TRAF6 which serves as convergence point for TLR-mediated MyD88-dependent and independent pathways leading to NF-κB activation, mediate both early and late phase of NF-κB activation by cooperating with MyD88 and TRIF, respectively [32]. Abrogation of NF-κB activation in cells transfected with DN–TRAF6 construct and TRIF inhibitory peptide, further confirmed a MyD88-independent and TRIF-dependent TLR4-mediated NF-κB activation.
Computational modeling of the two TLR4-dependent signaling pathways suggests that NF-κB activation through the MyD88-independent TRIF-dependent pathway occurs later than activation by the MyD88-dependent pathway [28]. MyD88-independent pathway requires IRF3-dependent TNFα expression to activate NF-κB and the time required for this TNFα synthesis establishes the delay [28]. Activation of NF-κB through the canonical TNFR-mediated pathway, which is a faster event [28], possibly compensates for the time delay required by MyD88-independent TRIF-dependent pathway to establish delayed activation of NF-κB in glioma cells. It is tempting to speculate that synergy between TLR4–NF-κB axis and canonical TNFα signaling-induced NF-κB sustains a feed-forward loop that maintains elevated NF-κB levels in glioma cells. The important finding of our study is that NF-κB integrates TNFα and TLR4 signals to promote pro-survival advantage and inflammatory response.
Induction of immunomodulatory cytokine IFNβ is dependent on IRF3 activation [43] and MyD88-independent TLR4 signaling activates IRF3 to subsequently trigger IFN-β [42, 44]. TLR4-induced IFN-β acts in an autocrine manner to activate the JAK-STAT pathway and expression of IFN-inducible and STAT-dependent genes [42, 45]. TLR4 affected TNFα-induced cytokines and chemokines differentially. This could be due to the fact that IFN-β does not affect NF-κB regulated genes IFN-γ-IP-10 and MCP-1 but negatively regulates IL-8 expression in glioma [46]. As TNFα-induced IFNβ-STAT1 pathway promotes a feed-forward cycle of inflammatory response [34], it is likely that TLR4-dependent IFNβ modulates inflammatory response in glioma.
Importantly, TLR4 regulates TNFα-induced increase in inflammatory cytokines and chemokines. We have demonstrated that HIF-1α-IL-1β feed-forward loop maintains persistently elevated IL-1β level [11] and that TNFα induces increased IL-6, IL-8 and MCP-1 release in glioma cells [47]. The chemokine IL-8 associated with glioma progression is induced in response to hypoxia and cytokines [48]. While IL-10 possibly contributes to glioma progression by suppressing immune response [49], IL-12 has been shown to have antitumor effects in a murine glioma model [50]. The chemokine MCP-1 recruits microglial cells to the site of glioma growth and increases tumor growth and neoangiogenesis [51]. It is likely that TNFα-induced TLR4-dependent production of both immunestimulatory and immunesuppressive mediators possibly negate the regulatory role of each other to prevent generation of an effective antitumoral immune response.
This study has highlighted for the first time the existence of TLR4–AKT–HIF-1α axis alongside the NF-κB–TLR4 loop in glioma cells. It is known that TLR4-dependent HIF-1α elevates cytokines associated with LPS-induced sepsis [21]. This, coupled with our finding that HIF-1α maintains persistently elevated IL-1β through an IL1β–HIF-1α autocrine loop [11] and our current identification of HIF-1α as an integral component of TLR4 signaling, further establishes HIF-1α as a crucial link between inflammatory and oncogenic components in GBM.
As HIF-1α is considered to be one of the most important anticancer target [14], elucidation of its involvement in signaling events emanating from TLR4, may provide new directions for therapeutic strategy for GBM. Taken together, our data clearly demonstrates that TNFα stimulated TLR4 drives inflammatory and oncogenic signals in glioma cells by triggering crosstalk between several signaling cascades. Stimulating TLRs to induce antitumor response is currently being considered as an attractive anticancer strategy [52]. However, the inability of TLR4 stimulation to elicit an antitumor immune response in murine glioma model [52] could have resulted from the fact that TLR4 signaling by itself triggers inflammatory response that possibly induces immune tolerance. Therefore, better understanding of TLR-induced inflammation in glioma will be crucial towards deciphering its role in antitumor immune response.
References
Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G (2008) Cancers take their Toll—the function and regulation of Toll-like receptors in cancer cells. Oncogene 27:225–233
Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, Visintin I, Rutherford T, Mor G (2006) TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res 66:3859–3868
Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M, Whiteside TL (2009) TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene 28:4353–4363
Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL (2009) Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res 69:3105–3113
He W, Liu Q, Wang L, Chen W, Li N, Cao X (2007) TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol 44:2850–2859
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Theofilopoulos AN, Baccala R, Beutler B, Kono DH (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336
Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH (2010) Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci USA 107:222–227
Nozell S, Laver T, Moseley D, Nowoslawski L, De Vos M, Atkinson GP, Harrison K, Nabors LB, Benveniste EN (2008) The ING4 tumor suppressor attenuates NF-kappaB activity at the promoters of target genes. Mol Cell Biol 28:6632–6645
Dong J, Jimi E, Zeiss C, Hayden MS, Ghosh S (2010) Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev 24:1709–1717
Sharma V, Dixit D, Koul N, Mehta VS, Sen E (2011) Ras regulates interleukin-1beta-induced HIF-1alpha transcriptional activity in glioblastoma. J Mol Med 89(2):123–136
Semenza GL (2002) HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 8:S62–S67
Gupta SC, Sundaram C, Reuter S, Aggarwal BB (2010) Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta 1799:775–787
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Gupta SC, Kim JH, Prasad S, Aggarwal BB (2010) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29:405–434
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L (2003) IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 17:2115–2117
Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG (2005) Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 7:134–153
Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL (2000) Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 88:2606–2618
Goto Y, Arigami T, Kitago M, Nguyen SL, Narita N, Ferrone S, Morton DL, Irie RF, Hoon DS (2008) Activation of Toll-like receptors 2, 3, and 4 on human melanoma cells induces inflammatory factors. Mol Cancer Ther 7:3642–3653
Sato Y, Goto Y, Narita N, Hoon DS (2009) Cancer cells expressing Toll-like receptors and the tumor microenvironment. Cancer Microenviron 2(Suppl 1):205–214
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V (2007) Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 178:7516–7519
Chen NJ, Chio II, Lin WJ, Duncan G, Chau H, Katz D, Huang HL, Pike KA, Hao Z, Su YW, Yamamoto K, de Pooter RF, Zuniga-Pflucker JC, Wakeham A, Yeh WC, Mak TW (2008) Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors. Proc Natl Acad Sci U S A 105:12429–12434
Muzio M, Ni J, Feng P, Dixit VM (1997) IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278:1612–1615
Biswas S, Gupta MK, Chattopadhyay D, Mukhopadhyay CK (2007) Insulin-induced activation of hypoxia-inducible factor-1 requires generation of reactive oxygen species by NADPH oxidase. Am J Physiol Heart Circ Physiol 292:H758–H766
Sharma V, Tewari R, Sk UH, Joseph C, Sen E (2008) Ebselen sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through two distinct pathways involving NF-kappaB downregulation and Fas-mediated formation of death inducing signaling complex. Int J Cancer 123:2204–2212
Sharma V, Joseph C, Ghosh S, Agarwal A, Mishra MK, Sen E (2007) Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol Cancer Ther 6:2544–2553
An H, Yu Y, Zhang M, Xu H, Qi R, Yan X, Liu S, Wang W, Guo Z, Guo J, Qin Z, Cao X (2002) Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology 106:38–45
Covert MW, Leung TH, Gaston JE, Baltimore D (2005) Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 309:1854–1857
Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, Hazeki O, Kitazaki T, Iizawa Y (2006) A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol 69:1288–1295
Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ (2008) Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science 321:259–263
Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A (2009) MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem 284:24192–24203
Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol 171:4304–4310
Wang H, Garcia CA, Rehani K, Cekic C, Alard P, Kinane DF, Mitchell T, Martin M (2008) IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-beta. J Immunol 181:6797–6802
Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB (2008) TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol 9:378–387
Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ (2003) Toll-like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J Biol Chem 278:22506–22512
Kuhlicke J, Frick JS, Morote-Garcia JC, Rosenberger P, Eltzschig HK (2007) Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS One 2:e1364
Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M (2008) NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453:807–811
Vivarelli MS, McDonald D, Miller M, Cusson N, Kelliher M, Geha RS (2004) RIP links TLR4 to Akt and is essential for cell survival in response to LPS stimulation. J Exp Med 200:399–404
Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A (2006) Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol Cancer Res 4:471–479
Ghosh S, Tewari R, Dixit D, Sen E (2010) TNFalpha induced oxidative stress dependent Akt signaling affects actin cytoskeletal organization in glioma cells. Neurochem Int 56:194–201
Kopp EB, Ghosh S (1995) NF-kappa B and rel proteins in innate immunity. Adv Immunol 58:1–27
Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13:816–825
Juang YT, Lowther W, Kellum M, Au WC, Lin R, Hiscott J, Pitha PM (1998) Primary activation of interferon A and interferon B gene transcription by interferon regulatory factor 3. Proc Natl Acad Sci U S A 95:9837–9842
Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN (2002) TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol 3:392–398
Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360
Nozell S, Laver T, Patel K, Benveniste EN (2006) Mechanism of IFN-beta-mediated inhibition of IL-8 gene expression in astroglioma cells. J Immunol 177:822–830
Tewari R, Sharma V, Koul N, Ghosh A, Joseph C, Hossain Sk U, Sen E (2009) Ebselen abrogates TNFalpha induced pro-inflammatory response in glioblastoma. Mol Oncol 3:77–83
Brat DJ, Bellail AC, Van Meir EG (2005) The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol 7:122–133
Huettner C, Paulus W, Roggendorf W (1995) Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am J Pathol 146:317–322
Hellums EK, Markert JM, Parker JN, He B, Perbal B, Roizman B, Whitley RJ, Langford CP, Bharara S, Gillespie GY (2005) Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol 7:213–224
Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, Weller M (2003) Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol 54:388–392
Grauer OM, Molling JW, Bennink E, Toonen LW, Sutmuller RP, Nierkens S, Adema GJ (2008) TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol 181:6720–6729
Acknowledgment
The work was supported by a research grant from the Department of Biotechnology–DBT (BT/PR12924/Med/30/235/09) to ES. We would like to thank Mr. Shanker Dutt Joshi for technical assistance and help with immunohistochemistry.
Disclosure
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Saurav Roy Choudhury and Sadashib Ghosh contributed equally to this work.
Rights and permissions
About this article
Cite this article
Tewari, R., Choudhury, S.R., Ghosh, S. et al. Involvement of TNFα-induced TLR4–NF-κB and TLR4–HIF-1α feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med 90, 67–80 (2012). https://doi.org/10.1007/s00109-011-0807-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0807-6