
Abstract
Limbic-predominant age-related TDP-43 encephalopathy (LATE) is characterized by the accumulation of TAR-DNA-binding protein 43 (TDP-43) aggregates in older adults. LATE coexists with Lewy body disease (LBD) as well as other neuropathological changes including Alzheimer’s disease (AD). We aimed to identify the pathological, clinical, and genetic characteristics of LATE in LBD (LATE-LBD) by comparing it with LATE in AD (LATE-AD), LATE with mixed pathology of LBD and AD (LATE-LBD + AD), and LATE alone (Pure LATE). We analyzed four cohorts of autopsy-confirmed LBD (n = 313), AD (n = 282), LBD + AD (n = 355), and aging (n = 111). We assessed the association of LATE with patient profiles including LBD subtype and AD neuropathologic change (ADNC). We studied the morphological and distributional differences between LATE-LBD and LATE-AD. By frequency analysis, we staged LATE-LBD and examined the association with cognitive impairment and genetic risk factors. Demographic analysis showed LATE associated with age in all four cohorts and the frequency of LATE was the highest in LBD + AD followed by AD, LBD, and Aging. LBD subtype and ADNC associated with LATE in LBD or AD but not in LBD + AD. Pathological analysis revealed that the hippocampal distribution of LATE was different between LATE-LBD and LATE-AD: neuronal cytoplasmic inclusions were more frequent in cornu ammonis 3 (CA3) in LATE-LBD compared to LATE-AD and abundant fine neurites composed of C-terminal truncated TDP-43 were found mainly in CA2 to subiculum in LATE-LBD, which were not as numerous in LATE-AD. Some of these fine neurites colocalized with phosphorylated α-synuclein. LATE-LBD staging showed LATE neuropathological changes spread in the dentate gyrus and brainstem earlier than in LATE-AD. The presence and prevalence of LATE in LBD associated with cognitive impairment independent of either LBD subtype or ADNC; LATE-LBD stage also associated with the genetic risk variants of TMEM106B rs1990622 and GRN rs5848. These data highlight clinicopathological and genetic features of LATE-LBD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
TAR-DNA-binding protein 43 (TDP-43) proteinopathy is characterized by abnormally phosphorylated, aggregated, cleaved, and mislocalized TDP-43 protein in the central and peripheral nervous systems [31, 39]. Although TDP-43 proteinopathy was first discovered in cases with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD-TDP) [39], it is also frequently observed in older adults in a distinct distribution called limbic-predominant age-related TDP-43 encephalopathy (LATE) [19, 35, 42]. LATE neuropathological change (LATE-NC) frequently coexists with other neurodegenerative diseases including Alzheimer’s disease (AD) [22, 41] and Lewy body disease (LBD) [2, 11, 32]. LATE-NC in AD (LATE-AD) is well characterized [39] and starts in the amygdala spreading to other regions in a stereotypical manner [15, 16]. LATE-AD is associated with AD neuropathologic change (ADNC), hippocampal sclerosis, brain atrophy, and cognitive impairment [1, 3, 6, 13, 29, 30, 34]. In particular, tau pathologies such as neurofibrillary tangles are strongly associated with LATE-NC [11, 14, 17, 43, 46]. LATE in LBD (LATE-LBD) is also associated with hippocampal sclerosis and the limbic-predominant subtype of LBD [2, 3, 22, 32, 41]. Some previous studies have indicated the possibility that pathological tau or α-synuclein influences the occurrence of TDP-43 pathology [7, 11, 26, 43]. Moreover, patients with mixed pathologies of tau, amyloid β, α-synuclein, and TDP-43 show more severe cognitive decline [18], suggesting the importance of co-pathologies in clinicopathological feature. Genetic analyses revealed that single-nucleotide polymorphisms (SNPs) associated with increased risk for FTLD-TDP, such as loci in transmembrane protein 106B (TMEM106B) [2, 12, 36] and progranulin (GRN) [12, 36], are also associated with both LATE-AD and LATE-LBD. However, it is unknown whether LATE-LBD has distinct characteristics in distribution or morphology compared with LATE-AD and if LATE-LBD is associated with cognitive impairment independent of LBD subtype or ADNC. Here, we identify morphological and distribution differences between LATE-LBD and LATE-AD and propose a staging of LATE-LBD. In addition, we report the clinical relevance of LATE-LBD independent of LBD subtype and ADNC and identify that genetic risk variants for FTLD-TDP, but not for Lewy body dementia, are associated with LATE-LBD stage.
Materials and methods
Participants
Demographic and neuropathological data were obtained for autopsy cases in the Integrated Neurodegenerative Disease (INDD) database [45, 50] at the Center for Neurodegenerative Disease Research (CNDR) from 1985 to 2021. (Table 1). Out of 1997 autopsy cases, the cases with primary neuropathological diagnosis of ALS, FTLD, Pick’s disease, argyrophilic grain disease, corticobasal degeneration, progressive supranuclear palsy, multiple system atrophy, Creutzfeldt–Jakob disease, Down syndrome, and fetal brain were excluded. After their exclusion, 1,061 cases were grouped into four cohorts by LBD subtype and ADNC according to 3rd and 4th McKeith criteria [23, 24] and National Institute on Aging and the Alzheimer’s Association (NIA-AA) guidelines [27, 28]: (1) LBD cohort, with cases that had more severe LBD than ADNC; (2) AD cohort, with cases that had “No” Lewy pathology with “Intermediate” or “High” ADNC; (3) LBD + AD cohort, with cases that had equivalent LBD and ADNC progression, or cases that had more advanced ADNC than LBD; and (4) Aging cohort, with cases that had “No” Lewy pathology with “Low” or “Not” ADNC (Fig. 1a). Out of 1,061 cases, a subset of patients had available Mini-Mental State Exam (MMSE) scores (n = 338–507) and genetic risk variant data (n = 770–987).

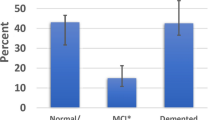
Comparison of LATE-LBD, LATE-AD, LATE-LBD + AD, and Aging. (a) The four cohorts examined for LATE (LBD, AD, LBD + AD, and Aging) and the screening method. (b) The ratio of LATE in each cohort. (c) Age dependency of LATE-LBD (P < 0.001), LATE-AD (P < 0.001), LATE-LBD + AD (P = 0.002), and Pure LATE (P = 0.004). (d) Association between LATE and LBD subtype (LATE-LBD, P = 0.012; LATE-LBD + AD, P = 0.314). (e) Association between LATE and ADNC (LATE-LBD, P = 0.002; LATE-AD, P = 0.002; LATE-LBD + AD, P = 0.662). Multiple logistic regression analysis was used with covariates of age at death and sex. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: AD Alzheimer’s disease; ADNC Alzheimer’s disease neuropathologic change; Amy amygdala; Ang angular gyrus; CA cornu ammonis; CB cerebellum; Cing cingulate gyrus; DG dentate gyrus; EC entorhinal cortex; LATE limbic-predominant age-related encephalopathy; LBD Lewy body disease; LN lentiform nucleus; MB midbrain; Med, medulla; MF middle frontal gyrus; OC occipital cortex; OFC orbitofrontal cortex; PAC periamygdaloid cortex; SMT superior/middle temporal gyrus; Sub subiculum; Th thalamus
Neuropathology and pathological analysis
Sixteen regions are routinely examined by neuropathologists (EBL and JQT) as described previously [45]. For TDP-43 staining, the orbitofrontal cortex region was added. The antibodies used in this study are described in Supplementary Table 1, online resource. Antigen retrieval using 88% formic acid was used [38]. The neuropathological changes were rated using a semi-quantitative scale (0, no changes; 0.5, rare; 1, mild; 2, moderate; 3, severe) as described previously [45]. The cases having neuropathological changes of 1, 2, and 3 were considered as “positive” in this study. LATE-NC in amygdala and hippocampal regions were used for screening of LATE. TDP-43 neuronal cytoplasmic inclusions (NCIs) and TPD-43 fine neurites in the amygdala, periamygdaloid cortex, dentate gyrus, cornu ammonis (CA), subiculum, entorhinal cortex, midbrain, pons, medulla, cingulate gyrus, orbitofrontal cortex, thalamus, lentiform nucleus, superior/middle temporal gyrus, middle frontal gyrus, angular gyrus, occipital gyrus, and cerebellum were further examined (Fig. 1a). The images were captured using Observer 7 (Zeiss), TCS SP8 WLL Confocal with STED 3X (Leica), or PANNORAMIC 250 (3DHISTECH) and analyzed using HALO (Indica Labs). To assess the association between LATE and patient profiles such as age at death, sex, disease duration, brain weight, hippocampal sclerosis, LBD subtype, and ADNC, multiple logistic regression analysis was used for LATE( +) or LATE(−) with covariates of age at death and sex.
LATE staging
Conditional probability analysis was applied for LATE-LBD stage, LATE-AD stage, and LATE-LBD + AD stage as previously reported [16, 21]. Briefly, if the number of cases in which the region X was positive and the region Y was negative (X + , Y −) and the number of cases in which the region X was negative and the region Y was positive (X − , Y +) were equally likely, those two regions were considered to be in the same stage. In contrast, if the number of cases with (X + , Y −) and the number of cases with (X − , Y +) were not equally likely, those two regions were considered to be in different stages. We used McNemar’s test to assess the evidence against the null hypothesis that (X + , Y −) and (X − , Y +) were equally likely and therefore X and Y were part of the same stage. Discordance was compared pairwise across all regions. Midbrain, pons, and medulla regions were combined as “brainstem” region to account for missing regions in some cases.
Cognitive analysis
Clinical diagnosis was prospectively determined for most of the cohorts using established criteria from consensus meetings in various clinical cores from the Penn Alzheimer Disease Research Center, Penn Frontotemporal Degeneration Center, and National Institute on Aging Penn U19 Center [45, 50]. The MMSE score [9] assessed at the time point closest to death was selected from the INDD database [45, 50] and used to assess antemortem global cognitive functioning [9]. MMSE data for cross-sectional cognitive analysis were available for 130 patients in the LBD cohort, 136 patients in the AD cohort, and 175 patients in the LBD + AD cohort (Supplementary Table 2, online resource). MMSE data for longitudinal cognitive analysis were available for 130 patients in the LBD cohort, 168 patients in the AD cohort, and 209 patients in the LBD + AD cohort (Supplementary Table 3, online resource). Factors that might also influence MMSE were included in the model: LBD subtype, ADNC, age at MMSE, years of education, and interval from MMSE to death. Analyses collapsed LATE across stages 3 and 4 to increase the sample size.
Genetic analysis
DNA was available in 988 cases from among 1,061 autopsy-confirmed cases. APOE isotype (E2, E3, or E4) was examined in 988 cases. Based on previous literature linking genetic variants to LATE-NC, we focused on TMEM106B rs1990622 (n = 987) [2, 48] and GRN rs5848 (n = 928) [8, 25, 36]. We also analyzed APOE rs769449 (n = 771) and TMEM175 rs6599388 (n = 770), which have been reported as genetic risk variants for Lewy body dementia [5]. Patient demographics are described in Supplementary Table 4, online resource. Genotype data were obtained from multiple datasets from either GWAS, a multiplex SNP panel, PANDoRA [25], which includes rs1990622 and rs5848, or Taqman assays (ThermoFisher, CA or Applied Biosystems, Foster City, CA) [48]. In 58 cases rs1990622 genotype was not obtained, and genotype at the highly linked proxy rs1468803 (D’ = 0.9835 R2 = 0.9633 in EUR population) was determined by Illumina Infinium Global Screening Array-24 v2.0 BeadChip (Illumina, San Diego, CA). Codominant, major, and minor allele dominant models are presented.
Statistical analysis
All statistical analyses were performed using R-4.0.5 or GraphPad Prism 7. For demographic analysis, multiple logistic regression analysis was used with covariates of age at death and sex. Statistical significance was set at P < 0.05. For pathological analyses, Wilcoxon tests compared differences between two independent groups and one-way ANOVA with post hoc Tukey test was used to compare three or more variables. Statistical significance was set at P < 0.05. For conditional probability analysis for LATE-LBD staging, LATE-AD staging, and LATE-LBD + AD staging, McNemar’s test was used to assess the evidence against the null hypothesis. We used P < 0.01 as a conservative value to determine whether there was sufficient evidence to reject the null hypothesis that two regions were part of the same stage. For cognitive analysis, Wilcoxon rank sum test was used to compare demographic characteristics in LBD patients between LATE( +) and LATE(−) and Kruskal–Wallis rank sum test was used to compare the interval from final MMSE to death between LATE( +) and LATE( −) in LBD, AD, and LBD + AD cohorts. Analysis of covariance (ANCOVA) using Type II sum of squares was used to compare final MMSE performance across LATE ( ±) or LATE stage with covariates of LBD subtype, ADNC, age at MMSE, years of education, and interval from the age at last MMSE measure to death. Statistical significance was set at P < 0.05. For the longitudinal study of cognitive decline, a liner mixed effects model was applied for the MMSE scores. Cohort, LATE(+ / −), interval from MMSE to death, age at onset, and education were included as fixed effects, and the interaction of Cohort, LATE(+ / −), and interval from MMSE to death was assessed in the model. For genetic analyses, multiple logistic regression analysis was used to determine the association between genotypes and LATE-LBD (+ / −) with covariates of age at death and sex. Ordinal logistic regression generated odds ratios (OR) to determine the association between genotypes and LATE-LBD stage (0, 1, 2, 3, 4) with covariates of age at death and sex. We did not find evidence against the proportional odds assumption. The statistical tests are two-sided. Statistical significance was set at P < 0.05.
Results
Comparison of LATE between LBD, AD, LBD + AD, and aging
Table 1 describes the demographics and characteristics of the patients in the cohorts. Cohorts were classified as described in Fig. 1a, resulting in the identification of 66 LATE-LBD, 95 LATE-AD, 152 LATE-LBD + AD, and 7 Pure LATE (Table 1). The frequency of LATE was the highest in LBD + AD (42.8%) followed by AD (33.7%), LBD (21.1%), and aging (6.3%) (Fig. 1b). Age at death was significantly associated with LATE in all cohorts (LBD, OR = 1.08, P < 0.001; AD, OR = 1.06, P < 0.001; LBD + AD, OR = 1.04, P = 0.002; Aging, OR = 1.15, P = 0.004) (Table 1 and Fig. 1c). LBD subtype was significantly associated with LATE in LBD (Diffuse or Neocortical > Transitional or Limbic > Brainstem Predominant, OR = 1.45, P = 0.012) but not in LBD + AD (P = 0.314) (Table 1 and Fig. 1d). Likewise, ADNC was significantly associated with LATE in LBD (Intermediate > Low > Not, OR = 2.03, P = 0.002) and AD (High > Intermediate, OR = 3.00, P = 0.002) but not in LBD + AD (P = 0.662) (Table 1 and Fig. 1e).
Distributional and morphological differences between LATE-LBD and LATE-AD
To see the pathological characteristics of LATE-LBD, we compared LATE-NC in LATE-LBD with that in LATE-AD. The amygdala and hippocampus were frequently affected in both LATE-LBD and LATE-AD (LATE-LBD, ~ 98% of cases with amygdala LATE-NC and ~ 44% with hippocampus LATE-NC; LATE-AD, ~ 96% of cases with amygdala LATE-NC, ~ 72% with hippocampus LATE-NC). In the hippocampus, however, there were distributional and morphological differences between LATE-LBD and LATE-AD. LATE-LBD had more TDP-43 NCIs than LATE-AD in the CA3 region (CA3, P = 0.033), while LATE-AD had more TDP-43 NCIs than LATE-LBD in the CA1 region and subiculum (CA1, P < 0.001; Subiculum, P = 0.002) (Fig. 2a, b, c). In the CA1 region, even though TDP-43 NCIs were more common in LATE-AD than LATE-LBD (P < 0.001) (Fig. 2d), TDP-43 fine neurites were more abundant in LATE-LBD than LATE-AD (P = 0.005) (Fig. 2e).

Morphological and distributional difference of LATE-NC between LATE-LBD and LATE-AD. (a, b) Representative images of LATE-LBD (a) and LATE-AD (b). (c) The ratio of TDP-43 neuronal cytoplasmic inclusions (NCIs) in dentate gyrus (DG), CA3, CA2, CA1, and subiculum. LATE-LBD shows more NCIs than LATE-AD in the CA3 while LATE-AD shows more NCIs than LATE-LBD in the CA1 and subiculum (DG, P = 0.487; CA3, P = 0.033; CA2, P = 0.376; CA1, P < 0.001; Subiculum, P = 0.002). (d) The number of TDP-43 NCIs/mm2 in CA1 of LATE-LBD and LATE-AD. The number of NCIs/mm2 in the CA1 of LATE-AD is 2.5 times higher than LATE-LBD (P < 0.001). (e) The % area of TDP-43 fine neurites (FNs) in the CA1 of LATE-LBD and LATE-AD. The % of TDP-43 FNs in the CA1 of LATE-LBD is 5.9 times higher than LATE-LBD (P = 0.005). (f) Representative images of immunofluorescence using TDP-43 p409/410 and syn303 antibodies, with nucleus staining with DAPI. Bars show 500 µm and 50 µm (enlarged images). (g) The % area of TDP-43 p409/410 positive FNs in each region of hippocampus. The TDP-43 p409/410 positive FNs are most observed in the CA1 region (CA1 vs DG, P < 0.001; CA1 vs. CA3, P < 0.001; CA1 vs. CA2, P = 0.043; CA1 vs. Subiculum, P < 0.001). (h) The % colocalized area of TDP-43 p409/410 and syn303 in each region of hippocampus. The TDP-43 p409/410 positive FNs are most observed in the CA2 region (CA2 vs. DG, P < 0.001; CA2 vs. CA3, P < 0.001; CA2 vs CA1, P < 0.001; CA2 vs. Subiculum, P < 0.001). Mann–Whitney U tests compared differences between two independent groups and one-way ANOVA with post hoc Tukey test was used to compare three or more variables. Statistical significance was set at P < 0.05 level
To examine LATE-NC in more detail in the hippocampus of LATE-LBD, we performed double immunofluorescence using phosphorylated TDP-43 (pSer409/410) and phosphorylated α-synuclein (syn303) antibodies (Fig. 2f). Some TDP-43 NCIs were colocalized with phosphorylated α-synuclein, which were mainly observed in CA3 and CA2. Likewise, TDP-43 fine neurites were colocalized with phosphorylated α-synuclein. Although the amount of TDP-43 fine neurites was the most in CA1 (Fig. 2f, g), the TDP-43 fine neurites that were colocalized with phosphorylated α-synuclein were most frequently observed in CA2 (Fig. 2f, h). To examine the composition of these TDP-43 fine neurites, we performed immunohistochemistry using a panel of antibodies which bind to different epitopes of TDP-43 (Supplementary Fig. 2a, online resource). Notably, TDP-43 fine neurites were stained by C-terminal TDP-43 antibodies that recognize TDP-43 with known C-terminal truncations but not N-terminal TDP-43 antibodies, suggesting these fine neurites are composed of C-terminal fragments of truncated TDP-43 (Supplementary Fig. 2b, online resource). The possibility that a particular epitope is buried because of the conformation of TDP-43 aggregates was excluded by pre-treating the tissue with 88% formic acid for antigen retrieval [31].
We also studied the LATE-NC associated with neurofibrillary tangles, which is called as TDP-43 type β [14], in LATE-LBD, LATE-AD, and LATE-LBD + AD (Supplementary Fig. 2, online source). The amount of LATE-NC colocalized with neurofibrillary tangles stained with PHF1 antibody was significantly associated with the amount pathology of neurofibrillary tangles and neuropil threads (r = 0.49, P = 0.009). This association was not different between LATE-LBD, LATE-AD, and LATE-LBD + AD (F(2, 24) = 0.52, P = 0.602).
LATE stage in LBD, AD, and LBD + AD
We assessed the frequency of LATE-NC across brain regions (Fig. 3). We did not find LATE-NC in the occipital cortex or cerebellum. We ordered each region according to the frequency of LATE-NC (Fig. 4a, and Supplementary Fig. 3a and c, online resource) and we then performed conditional probability analyses to group brain regions into stages of disease (Fig. 4b, Supplementary Fig. 3b and d, online resource). These analyses revealed the likely sequential spread of LATE-LBD (Fig. 4c). LATE-LBD appears to spread from the amygdala and the periamygdaloid cortex (stage 1) to dentate gyrus, CA, subiculum, and the entorhinal cortex (stage 2), then to the brainstem and cingulate gyrus (stage 3) and finally to the orbitofrontal cortex, thalamus, lentiform nucleus, temporal lobes, frontal lobes, and angular gyrus (stage 4). Table 2 describes the frequency of regional TDP-43 pathologies in each stage of LATE-LBD. The frequency pattern of TDP-43 in AD was consistent with the LATE-AD stage as previously described by Josephs et al. [16] with one minor exception: while Josephs et al. staged LATE-AD as six stages, the frequency of LATE-NC in the brainstem, subcortical, and frontal and parietal regions were not significantly separated in LATE-AD in our cohort and we, thus, staged LATE-AD as five stages (Supplementary Fig. 3a and b, and Supplementary Table 5, online resource).

Representative images of LATE-NC in each region of LATE-LBD. Representative images of LATE-NC in the amygdala (a), periamygdaloid cortex (b), dentate gyrus (c), CA (d), subiculum (e), entorhinal cortex (f), midbrain (g), pons (h), medulla (i), cingulate gyrus (j), orbitofrontal cortex (k), thalamus (l), lentiform nucleus (m), superior/middle temporal gyrus (n), middle frontal gyrus (o), and angular gyrus (p). The density of LATE-NC is higher in the amygdala, periamygdaloid cortex, and hippocampus than other regions. Bar shows 20 µm

LATE-LBD stage. (a) The % of LATE-NC in each area of LATE-LBD. (b) Conditional probability of LATE-NC between brain regions. The numbers in red shows P < 0.01 and the numbers in blue shows P < 0.05 using McNemar’s test. (c) Sagittal brain images showing the progression pattern of LATE-NC in LATE-LBD (left) and LATE-AD (right). Insets show hippocampal images showing dentate gyrus, CA, subiculum, and entorhinal cortex. The numbers in LATE-LBD show LATE-LBD stage (0–4) and the numbers in LATE-AD show LATE-AD stage (0–5)
We found two differences in LATE-LBD and LATE-AD. First, in LATE-LBD, the frequency of LATE-NC in the dentate gyrus was similar to that of CA, subiculum, and entorhinal cortex and those were placed in the same stage (stage 2). In LATE-AD, in contrast, LATE-NC in the dentate gyrus was not observed without the co-occurrence of LATE-NC in the CA and entorhinal cortex and hence dentate gyrus was placed in stage 3. Second, LATE-NC in the brainstem was more prevalent than in the temporal lobe in LATE-LBD, which was the opposite of that observed in LATE-AD (Fig. 4c). We also assessed LATE stages in the LBD + AD cohort. The frequency pattern of LATE-NC in LBD + AD had features common to LATE-LBD and LATE-AD. Similar to LATE-AD, dentate gyrus pathology appears to be a later event than hippocampal CA and entorhinal pathology. However, similar to LATE-LBD, the frequency of LATE-NC in the brainstem appears to be an earlier event in LBD + AD, and the brainstem, allocortical, subcortical, and neocortical areas were not clearly separable into distinct stages.(Supplementary Fig. 3c and d, online resource).
Association of LATE-LBD with cognitive impairment
To determine whether there was an association between LATE and cognitive impairment in each cohort, we examined MMSE scores assessed at the time point closest to death. The median of the interval from MMSE to death, which affects the MMSE scores, was not significantly different between LATE( +) and LATE(-) in the LBD, AD, and LBD + AD cohorts (median is 2–3 years; LBD, P = 0.825; AD, P = 0.083; LBD + AD, P = 0.051, all cohorts, P = 0.084) (Supplementary Table 2, online resource). In all cohorts, LATE( +) was significantly associated with lower MMSE (F(1, 494) = 14.35, P < 0.001) (Supplementary Fig. 4a, online resources). In addition, the longitudinal cognitive decline was faster in LATE( +) cases than LATE( −) cases (β = 34.03, P < 0.001) (Supplementary Fig. 4b, online resources). In the LBD cohort, lower MMSE score was significantly associated with LATE-LBD (F(1, 106) = 9.59, P = 0.003) (Fig. 5a, Table 3). On the other hand, in the AD and LBD + AD cohorts, lower MMSE was significantly associated with ADNC (AD, F(1, 124) = 9.45, P = 0.002; LBD + AD, F(1, 164) = 5.15, P = 0.024) but not with LATE (AD, F(1, 124) = 0.73, P = 0.393; LBD + AD, F(1, 164) = 1.08, P = 0.300) (Fig. 5a and Supplementary Table 6 online resource). Because MMSE scores in the AD and LBD + AD cohorts may have reached a plateau, we separately analyzed the MMSE scores in the cases with intermediate (Braak III/IV) and high (Braak V/VI) Braak stages. With intermediate Braak stage, the LATE( +) cases in the LBD + AD cohort showed significantly lower MMSE scores than LATE( −) cases (F(1, 9) = 5.19, P = 0.049), but there was no difference between LATE( +) and LATE( −) cases in the AD cohort (F(1, 12) = 0.05, P = 0.822). At high Braak stage, all cases – LATE( +) or LATE( −) – had low MMSE scores in both the AD and LBD + AD cohorts (Supplementary Fig. 4c, online resource). We also assessed the effect of LATE on longitudinal cognitive decline in the LBD, AD, and LBD + AD cohorts. (Fig. 5b). LATE( +) was significantly associated with cognitive decline in all cohorts (β = 103.40, P < 0.001). The effect of LATE( +) on cognitive decline was greater in the LBD than AD or LBD + AD (AD vs. LBD, β = 97.37, P < 0.001; LBD + AD vs. LBD, β = 79.77, P = 0.002).

Association between LATE and cognitive impairment in LBD, AD, and LBD + AD. a The final MMSE scores of LATE( +) and LATE( −) in LBD, AD, and LBD + AD cohorts. LATE( +) shows significantly lower MMSE than LATE( −) in LBD (F(1106) = 9.59, P = 0.003) but not in AD (F(1124) = 0.73, P = 0.393) or in LBD + AD (F(1,164) = 1.08, P = 0.300). (b) Longitudinal study of cognitive decline by LATE( +) or LATE( −) in LBD, AD, and LBD + AD cohorts. LATE( +) was significantly associated with cognitive decline in all cohorts (β = 103.40, P < 0.001). The effect of LATE( +) on the cognitive decline was greater in the LATE-LBD than LATE-AD (β = 97.37, P < 0.001) or LATE = LBD + AD (β = 79.77, P = 0.002). (c) LBD cohort MMSE scores of LATE( +) and LATE( −) in each LBD subtype. MMSE scores were lower according to LBD subtype (diffuse or neocortical < transitional or limbic < brainstem predominant) and LATE( +) shows lower MMSE than LATE(-) in each LBD subtype (LATE (F (1106) = 9.59, P = 0.003; LBD subtype (F (1,106) = 3.38, P = 0.038). (d) MMSE score associated with LATE-LBD stage (F (3104) = 3.92, P = 0.011). Analysis of covariance (ANCOVA) using Type II sum of squares was used to compare MMSE performance across LATE ( ±) or LATE stage (0, 1, 2, 3/4) with covariates of LBD subtype, ADNC, age at MMSE, years of education, and interval from the age at MMSE to death. Statistical significance was set at P < 0.05 level
In the LBD cohort, advanced LBD subtype cases were associated with lower MMSE scores (diffuse or neocortical < transitional or limbic < brainstem predominant) and MMSE scores were lower in LATE( +) than LATE( −) in each LBD subtype (LATE, F(1, 106) = 8.58, P = 0.004; LBD subtype, F(2, 106) = 3.41, P = 0.037) (Fig. 5c, Table 3). In the LBD + AD cohort, however, neither LATE nor LBD subtype was associated with lower MMSE (LATE, F(1, 197) = 0.43, P = 0.515; LBD subtype, F(3, 107) = 0.56, P = 0.642) (Supplementary Fig. 4d and Supplementary Table 6 online source).
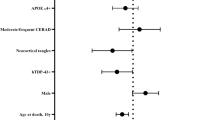
In the LBD cohort, lower MMSE was significantly associated with LATE-LBD stage and LBD subtype (LATE stage, F(3, 104) = 3,92, P = 0.011; LBD subtype, F(104, 2) = 3,27, P = 0.042) (Fig. 5d, Table 3). In the AD cohort, however, lower MMSE was significantly associated with ADNC (F(1, 153) = 11.47, P = 0.001) but not with LATE-AD stage (F(5, 153) = 0.60, P = 0.072). (Supplementary Fig. 4b and Supplementary Table 6, online source) (Table 4).
Genetic risk variants for LATE-LBD
To examine the genetic risk factors associated with LATE, we assessed TMEM106B rs1990622 and GRN rs5848, which are known risk factors for FTLD-TDP, and APOE rs769449 and TMEM175 rs6599388, which are associated with Lewy body dementia [5] (Supplementary Table 7, online resource). We first examined whether these genetic risk variants were associated with the presence or absence of LATE-NC. As reported previously [2, 35], in the setting of LBD, LATE( +) was more likely to have the TT TMEM106B rs199062 genotype compared to the LATE-LBD(−) group after controlling for age at death and sex (CC/TT, OR = 1.21, P = 0.009). In the minor allele dominant model, which compares major allele homozygotes to all other cases, the TT genotype of TMEM106B was also more likely to associate with LATE ( +) (CC + TC/TT, OR = 1.19, P = 0.001). Other genotypes did not associate with LATE-NC in LBD (P > 0.1). In the aging cohort, GRN was significantly associated with LATE in codominant (CC/CT, OR = 1.13, P = 0.028; CC/TT, OR = 1.17, P = 0.024) and minor allele dominant (CC + CT/TT, OR = 1.14, P = 0.008) models. None of the variants were associated with LATE-NC in the setting of AD.
We next examined the association between the genetic variants and the stage of LATE-LBD or LATE-AD (Table 4). In the LBD cohort, TMEM106B risk variants were associated with advanced LATE-LBD stage cases compared to less advanced LATE-LBD stage cases (in the codominant model, CC/TT, OR 3.36, P = 0.016; in the minor allele dominant model, CC + TC/TT, OR 2.84, P = 0.001). Likewise, the risk allele TT of GRN for FTLD-TDP was also associated with LATE-LBD stage in the major allele dominant model (CC + CT/TT OR = 2.56, P = 0.035). Unlike TMEM106b and GRN, the APOE rs769449 and TMEM175 rs6599388 variants were not significantly associated with LATE-LBD stage (P > 0.1). In the AD, none of four genetic variants associated with LATE-AD stage.
Discussion
In our study, we identified the characteristics of LATE-LBD, LATE-AD, and LATE-LBD + AD, and sufficient differences between LATE-LBD and LATE-AD were observed to warrant a distinct staging system for the distribution of LATE-NC in LBD.
The frequency of LATE was highest in the LBD + AD cases, suggesting co-pathologies of LBD and AD increase the development of LATE-NC. In these LBD + AD cases, the presence of LATE-NC was not associated with the severity of LBD or AD, suggesting that having co-pathologies of LBD and AD might increase the opportunity to develop additional pathology of LATE even though the severity of each co-pathology is small.
The primary differences between LATE-LBD and LATE-AD included the distinct neuropathological features of LATE-NC in the hippocampus and the different patterns of spread. Within the hippocampus, there were differences in the location of LATE-NC, as the CA3 region had more abundant TDP-43 NCIs in LATE-LBD while more NCIs were observed in the CA1 region in LATE-AD. Additionally, TDP-43 fine neurites were occasionally a conspicuous feature in LATE-LBD involving CA-to-subiculum regions. The sparse TDP-43 NCIs and abundant TDP-43 fine neurites in the CA1 region in LATE-LBD is consistent with a previous study [2]. Notably, some TDP-43 fine neurites were colocalized with Lewy neurites, suggesting that there may be a mechanistic link between TDP-43 and α-synuclein pathologies. Considering that α-synuclein aggregates have been reported to seed the C-terminal domain of TDP-43 monomers but not vice versa [7], the implication is that TDP-43 fine neurites may be induced by α-synuclein inclusions.
In addition, while the progression pattern of LATE-AD was similar to that described in previous studies [16], LATE-LBD showed a different pattern of spread within the hippocampal formation and an earlier involvement of the brainstem which becomes affected before neocortical regions. We speculate that these differences are due to the influence of tau versus α-synuclein pathology in AD versus LBD, respectively. Thus, in LATE-AD, the significant neurofibrillary tangle burden in the temporal lobe may correlate with the spread of TDP-43 into this region. In contrast, the significant Lewy pathology in the brainstem may correlate with the LATE-NC seen in LATE-LBD. Similarly, α-synuclein aggregates tend to form in the CA3 region of the hippocampus [4], which correlates with the tendency for increased TDP-43 inclusion burden in the CA3 region in LATE-LBD. Other studies have also indicated the interaction between TDP-43 and α-synuclein [11, 32, 44] or TDP-43 and tau [11, 14, 26, 43, 46]. The abundant neurofibrillary tangles in the CA/subiculum and the entorhinal cortex in LATE-AD may also propel LATE-NC in those areas, thereby separating involvement of CA/subiculum and entorhinal cortex (stage 2) from involvement of the dentate gyrus (stage 3), which is not the case for LATE-LBD where LATE-NC emerges equally frequently in those areas (stage 2). Indeed, Josephs et al. reported that TDP-43 immunoreactivity was often adjacent to or associated with neurofibrillary tangles, which they have designated TDP type-β [14].
Importantly, we show that the presence of LATE-NC in LBD is associated with cognitive impairment. Although neocortical LBs are highly significant correlates of cognitive impairment in LBD [33, 49], LATE( +) cases had lower MMSE scores than LATE(-) cases in LBD after controlling for LBD subtype and ADNC. LATE( +) cases also showed faster cognitive decline. These results suggest that LATE-NC in LBD may be an independent factor contributing to cognitive impairment as it is in other cohorts [35]. Furthermore, higher LATE-LBD stages also associated with lower MMSE scores, suggesting the severity and progression of LATE-LBD may be associated with worsening cognitive impairment, thereby validating the LATE-LBD staging scheme proposed here. Unlike LATE-LBD, LATE-AD stage did not associate with cognitive impairment in our cohort highlighting potential differences of LATE stage in LBD and AD. In the LBD + AD cohort, the cognitive decline was greater and the final MMSE score was much lower than both LATE( +) and LATE(-) groups in the LBD cohort, which is consistent with the previous report that the cases with LATE + LBD + AD co-pathology had a more aggressive clinical course and affected younger subjects [18]. Furthermore, unlike the LBD cohort, only ADNC, but not LATE or LBD subtype, associated with cognitive impairment at the closest point to death in LBD + AD cohort, suggesting ADNC is a stronger factor for cognitive impairment than the neuropathological changes of LATE or LBD.
TMEM106B rs1990622 is a genetic risk factor for FTLD-TDP [10, 48, 51], as well as for cognitive impairment [47] and hippocampal sclerosis [2, 36] in LBD. GRN rs5848 is also a genetic risk factor for FTLD-TDP [8] and hippocampal sclerosis in AD or aging cases [36]. In the current study, we determined whether these genetic variants were associated with either the occurrence (LATE(+ / −)) or spreading (LATE-LBD stage) of LATE-NC. In terms of the presence/absence of LATE-NC, only TMEM106B rs1990622 was associated with LATE in the LBD, whereas GRN rs5848 was associated with Pure LATE, which is consistent with a previous report [2]. We also observed that TMEM106B rs1990622 and GRN rs5848 were associated with LATE-LBD stage. The results suggest that both TMEM106B rs1990622 and GRN rs5848 are risk factors for the spread of LATE-NC in LBD, which may potentially represent the neuropathologic substrate for cognitive decline in LBD. In contrast with these two FTLD-TDP risk variants, Lewy body dementia risk variants APOE rs769449 and TMEM175 rs6599388 [5] did not associate with LATE-LBD.
This study has some limitations. First, the number of LATE-LBD cases with available cognitive profiles was relatively small for our LATE-LBD stage analysis. Therefore, to improve statistical validity, LATE stages 3 and 4 were combined in the clinical analyses. Future replication studies will be needed to confirm our results. Nonetheless, we note that neuropsychological testing data in autopsy-confirmed LBD is extremely rare and our findings evidence that LATE is clinically relevant in LBD. Second, only Lewy pathology and ADNC were considered as pathologies that may associate with LATE and cognitive impairment in this study. Other pathologies such as cerebrovascular disease and aging-related tau astrogliopathy could also be considered. Given that vascular factors influence cognitive impairment and are often observed in LATE, LBD, AD, and hippocampal sclerosis [20, 37, 40], more comprehensive analysis will likely demonstrate that there are heterogeneous pathologies that associated with LATE.
In summary, this study revealed distinct morphological and distributional characteristics of LATE in LBD. Furthermore, this study found an association between LATE-LBD and its progression pattern with cognitive impairment and genetic risk variants of TMEM106B rs1990622 and GRN rs5848. Therefore, the clinical phenotype in LBD may be the consequence of multiple heterogeneous underlying pathologies, including LATE-NC. These results suggest that clinical trials may need to consider the considerable degree of pathologic heterogeneity that drives cognitive dysfunction in Parkinson’s disease and dementia with Lewy bodies.
References
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. https://doi.org/10.1002/ana.21154
Aoki N, Murray ME, Ogaki K, Fujioka S, Rutherford NJ, Rademakers R et al (2015) Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol 129:53–64. https://doi.org/10.1007/s00401-014-1358-z
Besser LM, Teylan MA, Nelson PT (2020) Limbic predominant age-related TDP-43 encephalopathy (LATE): clinical and neuropathological associations. J Neuropathol Exp Neurol 79:305–313. https://doi.org/10.1093/jnen/nlz126
Braak H, Tredici K, Rüb U, Vos Rd, Steur E, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. https://doi.org/10.1016/S0197-4580(02)00065-9PMID-12498954
Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E et al (2021) Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 53:294–303. https://doi.org/10.1038/s41588-021-00785-3
de Flores R, Wisse LEM, Das SR, Xie L, McMillan CT, Trojanowski JQ et al (2020) Contribution of mixed pathology to medial temporal lobe atrophy in Alzheimer’s disease. Alzheimers Dement 16:843–852. https://doi.org/10.1002/alz.12079
Dhakal S, Wyant CE, George HE, Morgan SE, Rangachari V (2021) Prion-like C-terminal domain of TDP-43 and α-Synuclein interact synergistically to generate neurotoxic hybrid fibrils. J Mol Biol. https://doi.org/10.1016/j.jmb.2021.166953
Dickson DW, Baker M, Rademakers R (2010) Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis 7:170–174. https://doi.org/10.1159/000289231
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. https://doi.org/10.1016/0022-3956(75)90026-6
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC et al (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127:407–418. https://doi.org/10.1007/s00401-013-1239-x
Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K et al (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184:284–294. https://doi.org/10.1016/j.brainres.2007.09.048
Hokkanen SRK, Kero M, Kaivola K, Hunter S, Keage HAD, Kiviharju A et al (2020) Putative risk alleles for LATE-NC with hippocampal sclerosis in population-representative autopsy cohorts. Brain Pathol 30:364–372. https://doi.org/10.1111/bpa.12773
Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L et al (2017) Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol 16:917–924. https://doi.org/10.1016/s1474-4422(17)30284-3
Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB et al (2019) Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol 137:1–12. https://doi.org/10.1007/s00401-018-1951-7PMID-30604226
Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR et al (2014) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127:441–450. https://doi.org/10.1007/s00401-013-1211-9
Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L et al (2016) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131:571–585. https://doi.org/10.1007/s00401-016-1537-1
Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM et al (2015) TAR DNA-binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Ann Neurol 78:697–709. https://doi.org/10.1002/ana.24493
Karanth S, Nelson PT, Katsumata Y, Kryscio RJ, Schmitt FA, Fardo DW et al (2020) Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol 77:1299–1307. https://doi.org/10.1001/jamaneurol.2020.1741
Katsumata Y, Abner EL, Karanth S, Teylan MA, Mock CN, Cykowski MD et al (2020) Distinct clinicopathologic clusters of persons with TDP-43 proteinopathy. Acta Neuropathol 140:659–674. https://doi.org/10.1007/s00401-020-02211-0
Katsumata Y, Fardo DW, Kukull WA, Nelson PT (2018) Dichotomous scoring of TDP-43 proteinopathy from specific brain regions in 27 academic research centers: associations with Alzheimer’s disease and cerebrovascular disease pathologies. Acta Neuropathol Commun 6:142. https://doi.org/10.1186/s40478-018-0641-y
Kovacs GG, Xie SX, Robinson JL, Lee EB, Smith DH, Schuck T et al (2018) Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun 6:50. https://doi.org/10.1186/s40478-018-0552-y
McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J (2017) TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 27:472–479. https://doi.org/10.1111/bpa.12424
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D et al (2017) Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 89:88–100. https://doi.org/10.1212/WNL.0000000000004058
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65:1863–1872. https://doi.org/10.1212/01.wnl.0000187889.17253.b1PMID-16237129
McMillan CT, Toledo JB, Avants BB, Cook PA, Wood EM, Suh E et al (2014) Genetic and neuroanatomic associations in sporadic frontotemporal lobar degeneration. Neurobiol Aging 35:1473–1482. https://doi.org/10.1016/j.neurobiolaging.2013.11.029
Montalbano M, McAllen S, Cascio FL, Sengupta U, Garcia S, Bhatt N et al (2020) TDP-43 and Tau oligomers in Alzheimer’s disease, amyotrophic lateral sclerosis, and frontotemporal dementia. Neurobiol Dis 146:105130. https://doi.org/10.1016/j.nbd.2020.105130
Montine TJ, Monsell SE, Beach TG, Bigio EH, Bu Y, Cairns NJ et al (2016) Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer’s disease. J lzheimer’s Dementia Assoc 12:164–169. https://doi.org/10.1016/j.jalz.2015.07.492
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1–11. https://doi.org/10.1007/s00401-011-0910-3
Murray ME, Cannon A, Graff-Radford NR, Liesinger AM, Rutherford NJ, Ross OA et al (2014) Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 128:411–421. https://doi.org/10.1007/s00401-014-1302-2
Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA et al (2015) Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol 77:942–952. https://doi.org/10.1002/ana.24388
Nakamura-Shindo K, Sakai K, Shimizu A, Ishida C, Yamada M (2020) Accumulation of phosphorylated TDP-43 in the cytoplasm of Schwann cells in a case of sporadic amyotrophic lateral sclerosis. Neuropathology 40:606–610. https://doi.org/10.1111/neup.12673
Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE et al (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114:221–229. https://doi.org/10.1007/s00401-007-0261-2
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ et al (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. https://doi.org/10.1097/NEN.0b013e31825018f7
Nelson PT, Braak H, Markesbery WR (2009) Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68:1–14. https://doi.org/10.1097/NEN.0b013e3181919a48
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503–1527. https://doi.org/10.1093/brain/awz099
Nelson PT, Wang WX, Partch AB, Monsell SE, Valladares O, Ellingson SR et al (2015) Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 74:75–84. https://doi.org/10.1097/nen.0000000000000151
Neltner JH, Abner EL, Baker S, Schmitt FA, Kryscio RJ, Jicha GA et al (2014) Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 137:255–267. https://doi.org/10.1093/brain/awt318
Neumann M, Frick P, Paron F, Kosten J, Buratti E, Mackenzie IR (2020) Antibody against TDP-43 phosphorylated at serine 375 suggests conformational differences of TDP-43 aggregates among FTLD–TDP subtypes. Acta Neuropathol 140:645–658. https://doi.org/10.1007/s00401-020-02207-wPMID-32778941
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Raz L, Knoefel J, Bhaskar K (2016) The neuropathology and cerebrovascular mechanisms of dementia. J Cereb Blood Flow Metab 36:172–186. https://doi.org/10.1038/jcbfm.2015.164
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C et al (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141:2181–2193. https://doi.org/10.1093/brain/awy146
Robinson JL, Porta S, Garrett FG, Zhang P, Xie SX, Suh E et al (2020) Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 143:awaa219. https://doi.org/10.1093/brain/awaa219
Smith VD, Bachstetter AD, Ighodaro E, Roberts K, Abner EL, Fardo DW (2018) Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol 28:264–273. https://doi.org/10.1111/bpa.12505
Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG (2011) TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci 7:234–243. https://doi.org/10.7150/ijbs.7.234
Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL et al (2014) A platform for discovery: the University of Pennsylvania integrated neurodegenerative disease biobank. Alzheimers Dement 10:477-484.e471. https://doi.org/10.1016/j.jalz.2013.06.003
Tomé SO, Gomes LA, Li X, Vandenberghe R, Tousseyn T, Thal DR (2021) TDP-43 interacts with pathological τ protein in Alzheimer’s disease. Acta Neuropathol 141:795–799. https://doi.org/10.1007/s00401-021-02295-2
Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J et al (2019) TMEM106B effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol 85:801–811. https://doi.org/10.1002/ana.25486PMID-30973966
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. https://doi.org/10.1038/ng.536
Walker Z, Possin KL, Boeve BF, Aarsland D (2015) Lewy body dementias. Lancet (London, England) 386:1683–1697. https://doi.org/10.1016/S0140-6736(15)00462-6
Xie SX, Baek Y, Grossman M, Arnold SE, Karlawish J, Siderowf A et al (2011) Building an integrated neurodegenerative disease database at an academic health center. Alzheimers Dement 7:e84–e93. https://doi.org/10.1016/j.jalz.2010.08.233
Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA (2015) The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84:927–934. https://doi.org/10.1212/wnl.0000000000001313
Acknowledgements
DCK is currently affiliated with the John’s Hopkins University School of Medicine. We thank Manuela Neumann and Elisabeth Kremmer for providing the phosphorylation specific TDP-43 antibody TAR5P-1D3. We thank all of our colleagues in CNDR who helped with this study as well as all off the patients and their families.
Funding
MTU (201870008) and NU (201860169) report fellowships from the Japan Society for the Promotion of Science (JSPS). MTU report fellowships from the Uehara Memorial Foundation. KAQC reports Alzheimer’s Association Research Fellowship to Promote Diversity (AARF-D-619473) and the RAPID Program in Dementia (AARF-D-619473-RAPID). TFT (K23-NS11416-01A1), DAW (P30 AG010124), VMYL (T32-AG000255), EBL (P30 AG072979, P01 AG066597), and JQT (U19 AG062418, P50 NS053488) report grants from the US National Institute of Aging (NIA) or the US National Institute of Neurological Disorders and Stroke (NINDS) of National Institutes of Health (NIH) during this study. DJI reports grants from LBDA, Penn IOA. Data were contributed to this study by the Center on Alpha-synuclein Strains in Alzheimer Disease & Related Dementias at the University of Pennsylvania Perelman School of Medicine (U19 AG062418, Trojanowski JQ-PI) and the former Morris K. Udall Center at the University of Pennsylvania Perelman School of Medicine (P50 NS053488, Trojanowski JQ-PI).
Author information
Authors and Affiliations
Contributions
MTU, JLR, DJI, EBL, and JQT designed the study. MTU, JLR, JDM, SXX, NU, EBL, and JQT collected and analyzed pathological data. MTU, KAQC, SXX, and DJI collected and analyzed cognitive data. MTU, TFT, DCK, ES, SXX and VMVD analyzed genetic data. MTU drafted the article and all other authors assisted with revisions and approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
DAW reports grants from Merck, Biogen, and Avid/Eli Lilly. He has received personal fees from GE Healthcare, Functional Neuromodulation, and Neuronix. EBL and JQT are members of Acta Neuropathologica’s Editorial Board but were not involved in the editorial handling of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Uemura, M.T., Robinson, J.L., Cousins, K.A.Q. et al. Distinct characteristics of limbic-predominant age-related TDP-43 encephalopathy in Lewy body disease. Acta Neuropathol 143, 15–31 (2022). https://doi.org/10.1007/s00401-021-02383-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02383-3