
Abstract
Hippocampal sclerosis of the elderly (HpScl) and Alzheimer’s disease (AD), especially the limbic-predominant subtype (LP-AD), are amnestic syndromes that can be difficult to distinguish. To complicate matters, a subset has concomitant HpScl and AD (HpScl-AD). We examined a large cohort of autopsy-confirmed cases of HpScl, HpScl-AD, LP-AD, and typical AD to identify distinct clinical, genetic, and pathologic characteristics. HpScl cases were significantly older at death and had a substantially slower rate of cognitive decline than the AD subtypes. Genetic analysis revealed that the AD groups (AD, LP-AD, and HpScl–AD) were more likely to be APOE ε4 carriers. In contrast, the HpScl groups (HpScl and HpScl-AD) were more likely to exhibit genetic variants in GRN and TMEM106B that are associated with frontotemporal lobar degeneration. The HpScl groups had a high frequency of TDP-43 pathology that was most often Type A morphology and distribution, while typical AD and LP-AD had a significantly lower frequency of TDP-43 pathology that was most often Type B. These results suggest that HpScl and AD are pathologically and genetically distinct and non-synergistic neurodegenerative processes that present with amnestic dementia. Pure HpScl and HpScl with concomitant AD occur most often in elderly individuals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The differential diagnosis of late-onset amnestic dementia includes both Alzheimer’s disease (AD) and hippocampal sclerosis of the elderly (HpScl) [4, 31]. The neuropathologic diagnosis of AD requires the presence of neurofibrillary tangles (NFT) and senile plaques (SP) [16]. These lesions are usually abundant in hippocampus and neocortex; however, atypical distribution of AD pathology presenting with focal cortical atrophy and focal clinical presentations is increasingly recognized [1, 14]. AD can be neuropathologically classified into three subtypes based on the relative density of NFTs in the hippocampus and cortex: hippocampal-sparing AD, typical AD, and limbic-predominant AD (LP-AD) [17, 23]. In contrast to typical AD, which follows the Braak NFT staging scheme [5], LP-AD has disproportionately greater NFTs in the hippocampus than in the neocortex.
HpScl can be difficult to distinguish from LP-AD by neuroimaging and pathologic analysis given that the medial temporal lobe is severely affected in both diseases. HpScl is characterized by neuronal loss in the hippocampal CA1 sector and the subiculum that is disproportionate to the number of NFT, if any, in these regions [10]. Furthermore, HpScl of the elderly is not associated with epilepsy or history of hypoxic/ischemic injury. HpScl cases can be difficult to identify because the brains usually have confounding pathology, which often includes NFT and SP due to the advanced age of patients.
In an effort to distinguish late-onset amnestic dementias, we have investigated the neuropathologic, genetic, and clinical characteristics of a large cohort of autopsy-confirmed cases with typical AD, LP-AD, HpScl, and HpScl with concomitant AD (HpScl-AD). This study design also allowed us to determine if AD and HpScl are synergistic or separate disease processes.
Materials and methods
Study samples
Research protocols described in this report were approved by the Mayo Clinic Institutional Review Board. All brains were acquired and autopsied after informed consent by the legal next-of-kin. The Mayo Clinic Jacksonville brain bank was queried to identify the following study groups: typical AD (AD), limbic predominant AD (LP-AD), HpScl-AD, and HpScl of the elderly. The exclusion criteria for all cases were: unavailable tissue blocks, absence of NFT or SP counts, known pathogenic mutations, and history of epilepsy or seizure. All Alzheimer’s disease groups (AD, LP-AD, and HpScl-AD) were required to have a Braak stage ≥IV. The AD subtypes were classified using a mathematical algorithm that assesses NFT density in hippocampus and neocortex, as previously described [23]. Briefly, thioflavin-S fluorescent microscopy was used to assess density and distribution of NFTs in three neocortical regions and two hippocampal sectors in the original study population of 889 AD cases. LP-AD cases were identified if the hippocampal NFTs were disproportionately greater than cortical NFTs. Additionally, a ratio of hippocampal to cortical NFTs greater than the 75th percentile was used to avoid classifying mild cases. All HpScl (HpScl and HpScl-AD) had (1) disproportionate neuronal loss compared to NFT accumulation in the hippocampus (Fig. 1) and (2) TDP-43 neuropathology (Fig. 2). TDP-43 neuropathology was a requirement so that HpScl due to causes other than a neurodegenerative process would be excluded. Cases in the HpScl-AD group not only had a primary diagnosis of AD, but also neuronal loss disproportionate to extracellular NFT in the hippocampus. Cases in the HpScl group had no other significant coexisting pathology. Applying inclusion and exclusion criteria led to a sample size of 30 HpScl, 132 HpScl-AD, 151 LP-AD, and 807 typical AD. Study source can be found in Supplementary methods.

Histologic features by hippocampal sclerosis and AD subtype. H&E staining of the hippocampus (a, d, g, j) and especially the CA1 region (b, e, h, k) revealed neuronal loss. Thioflavin-S fluorescent microscopy of the CA1 region (c, f, i, l) was performed to detect senile plaques (SP) and neurofibrillary tangles (NFT). Cases with typical AD (a–c) usually have hippocampal neuron loss (a, b), many SPs and NFTs in hippocampal (c) and cortical regions. Cases with LP-AD (d–f) commonly have hippocampal neuron loss (d, e) with abundant NFTs relatively restricted to the hippocampus (f), without an apparent effect on SP distribution. Cases with HpScl-AD (g–i) have extensive hippocampal neuron loss (g, h), NFTs (i) and SPs in hippocampal and cortical regions. Cases with HpScl (j–l) have extensive hippocampal neuron loss mainly in the CA1 and subiculum regions (j, k), few SPs and NFTs in hippocampal (l) and cortical regions. Scale bar 1,000 microns (a, d, g, j), and 50 microns (b, c, e, f, h, i, k, l). AD Alzheimer’s disease, LP-AD limbic-predominant AD, HpScl hippocampal sclerosis, H&E hematoxylin&eosin, Thio-S Thioflavin-S

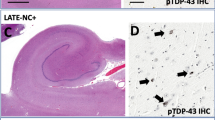
TDP-43 immunohistochemistry of the hippocampus was performed to detect TDP-43-positive inclusions and to determine the TDP-43 pathology classification. To illustrate differences across cases, the dentate fascia (a, d, g, j,), CA1 (b, e, h, k), and the superficial layer of the parahippocampal cortex (c, f, i, l) is shown. Of the subset of typical AD (a–c) and LP-AD (d–f) that are TDP-43 positive, there is a higher frequency of Type B TDP-43 pathology, characterized by a predominance of cortical NCIs (c, f) throughout cortical layers. Both NCIs and NIIs (a, center) were found in the dentate fascia granule cells (a, d). Neurofibrillary tangle-associated TDP-43 could be found in both typical AD (b) and LP-AD (e), with few NCIs observed. HpScl-AD (g–i) and HpScl (j–l) have a higher frequency of Type A TDP-43 pathology, characterized by cortical NCIs and neuritic pathology typically localized to superficial cortical layers (i, l). NCIs, NIIs, and rare neurites were found in the dentate fascia of HpScl-AD (g) and HpScl (j). Abundant neuritic pathology in the CA1 was observed in both HpScl-AD (h) and HpScl (k), as well as vascular TDP-43 (k, inset). Scale bar 50 microns. AD Alzheimer’s disease, LP-AD limbic-predominant AD, HpScl hippocampal sclerosis
Procedures
Gross and macroscopic neuropathologic assessment was performed by standardized procedures. Formalin-fixed, paraffin-embedded tissue samples from the primary motor and visual cortices, inferior parietal lobule, mid-frontal gyrus, superior temporal gyrus, amygdala, and posterior hippocampus were cut at 5 μm thickness, mounted on glass slides and stained with H&E. Thioflavin-S fluorescent microscopy was performed to evaluate SP (10× objective) and NFT densities (40× objective). Primitive, neuritic, and cored type plaques were included in the SP counts and were truncated at 50, which is twice the number required to meet Khachaturian’s criteria for AD diagnosis [20]. NFT distribution was assessed to determine Braak stage. Intracellular and extracellular NFT counts from two hippocampal regions (CA1 and subiculum) and three association cortices (inferior parietal, mid-frontal, and superior temporal) were used to determine AD subtypes. Additional sections previously underwent immunohistochemical staining and were processed using the DAKO Autostainer (DAKO Auto Machine Corporation, Carpinteria, CA, USA) with DAKO Envision+ HRP System. The posterior hippocampus was stained with an antibody that detects the 25 kDa C-terminal fragment of TDP-43 (a generous gift from Leonard Petrucelli, Mayo Clinic, FL, USA). Assessment of TDP-43 pathology was performed as previously described [2]. Cerebrovascular disease was assessed using a simple scheme proposed by Jellinger and Attems [18], as previously reported. Lewy body disease (LBD) pathology was assessed by use of immunohistochemistry [34], as previously described.
For genotyping, genomic DNA was extracted from frozen brain by standard procedures. Genotyping for MAPT H1/H2 (SNP rs1052554 A/G, A = H1, G = H2), APOE alleles (SNP rs429358 C/T and rs7412 C/T), GRN (SNP rs5848 C/T), and TMEM106B (SNP rs1990622 C/T) was performed using a Taqman SNP genotyping assay (Applied Biosystems, Carlsbad, CA, USA). Genotype calls were obtained with SDS v2.2 software (Applied Biosystems). Although there is overlap in the HpScl-AD group between previous genetic studies [9, 32], genetic information on the HpScl group has not been previously reported. Genotyping availability can be found in Supplementary methods.
Clinical reports were reviewed blind to pathologic diagnosis to collect education, family history, age of onset, disease duration, and Mini Mental State Examination (MMSE) scores. Family history was noted as positive if at least one first-degree relative had dementia. Age of onset was recorded as the age of initial cognitive abnormalities, as opposed to age of diagnosis. Disease duration was ascertained as the number of years elapsed between age of death and age of onset. Longitudinal decline was calculated as a slope of three or more MMSE scores, where the MMSE score was the dependent variable and elapsed years between testing and death were the independent variable. Antemortem clinical diagnosis of probable AD, possible AD and mild cognitive impairment was considered an amnestic diagnosis. Availability of clinical information can be found in Supplementary methods.
Statistical methods
All statistical analyses and graph generation were performed in SigmaPlot 11.0 (San Jose, CA, USA). A Kruskal–Wallis one-way analysis of variance on ranks was performed for group comparisons of continuous variables and a post hoc pairwise comparison was performed with Mann–Whitney rank sum test. A χ 2 test was performed for group comparisons of categorical data. Six multivariable logistic regression models were built for each combination of pathologic groups using the significant clinical and genetic variables from univariate analyses (Supplementary Table 1). Longitudinal decline on MMSE was not included given the limitation of sample size. An odds ratio <1 indicates that the independent variable (e.g., clinical or genetic feature) has a greater odds associated with the dependent pathologic “0” group. To facilitate interpretation of the odds, the inverse of the odds ratio (1/odds ratio) was calculated and used for interpretation in the “Results” section.
Results
Prior to implementing exclusion criteria, we assessed the frequency of HpScl in the Mayo Clinic brain bank in cases whose postmortem diagnosis was considered to underlie a dementing disorder (n = 5,074). There were 285 cases with concomitant HpScl or about 6 % of the “dementia” cases. A total of 30 autopsy-confirmed TDP-43 positive HpScl without significant coexisting pathology were included in the study. An additional 132 cases of HpScl with concomitant AD pathology were ascertained. A validated classification algorithm identified 807 typical AD and 151 LP-AD [23]. Of the 471 males and 649 females in the study, the LP-AD had the highest proportion of women (70 %; Table 1). The age at death was significantly later in HpScl (90 years) compared to typical AD (80 years, p < 0.001; Table 1), which is similar to previous findings [7, 27]. Both HpScl-AD and LP-AD had an average age of death of 86 years, midway between HpScl and typical AD. The median HpScl brain weight (1,160 g) was significantly higher than the AD groups (1,040 g, p < 0.001), with HpScl-AD having the lowest brain weight (1,010 g, p < 0.001; Table 1).
Neuropathologic findings
To determine if the study groups had distinct pathologic characteristics, we examined various pathologic parameters. We first investigated the characteristic lesions found in AD: NFTs and SPs (Fig. 1). The median Braak NFT stage for AD, LP-AD, and HpScl-AD was VI, while the HpScl Braak stage was II (Table 1). The NFTs in HpScl were sparse or absent in all regions examined. As expected, the LP-AD exhibited the highest NFT density within the hippocampal regions [17, 23]. NFT density within the cortical regions was highest in typical AD. The SP density within the cortical and hippocampal regions was similar across the typical AD, LP-AD, and HpScl-AD groups (Supplementary Table 2). In comparison, SP density in the HpScl group was significantly lower than all other groups (p < 0.001).
Focal hippocampal atrophy can be associated with a range of medical conditions including cardiovascular disease and traumatic brain injury [12]; therefore, to ensure atrophy was a result of a neurodegenerative process we only included HpScl and HpScl-AD that were positive for TDP-43. Although TDP-43 positivity was used as inclusion criteria for HpScl and HpScl-AD, we examined the relative frequency of cerebrovascular disease pathology across the groups given evidence of an association with certain forms of vascular pathology [28]. There were significant group-wise differences in cerebrovascular disease [18], with the highest frequency observed in HpScl cases and lowest frequency in typical AD cases; however, pairwise comparisons only revealed significant differences between typical and LP-AD (Table 1). LBD pathology was also found to significantly differ when a group-wise analysis was performed (Table 1), where HpScl had a much lower frequency of concomitant LBD pathology. Amygdala-predominant Lewy body disease was over-represented in LP-AD [17], but interestingly was far less in HpScl-AD compared to both typical AD and LP-AD.
TDP-43 pathology is strongly associated with HpScl and to a lesser extent with AD [2, 3, 19, 27], but the type of TDP-43 pathology has not been characterized between AD subtypes. Thus, we evaluated the TDP-43 frequency and subtype in our study groups (Fig. 2; Table 1). Only a subset of typical AD and LP-AD groups were TDP-43-positive (30 and 35 %, respectively) and usually exhibited a Type B [formerly Mackenzie type 3, with predominantly neuronal cytoplasmic inclusions (NCI) and sparse or no dystrophic neurites] pattern of TDP-43 distribution and morphology. In contrast, HpScl and HpScl-AD predominantly exhibited a Type A [formerly Mackenzie type 1, with NCI, dystrophic neurites and usually neuronal intranuclear inclusions (NII)] pattern. In total, these neuropathologic findings demonstrate that LP-AD neuropathology is more analogous to typical AD than the other study groups, and HpScl-AD exhibits features of both typical AD and HpScl.
Genetic analyses
We performed multiple genetic analyses to ascertain any differences in our study groups (Table 2). Assessment of MAPT revealed that LP-AD (71 %) had significantly more H1H1 homozygotes than typical AD (60 %, p = 0.022), but no other significant differences were found. We did not detect any APOE ε4 allele effects between the AD groups (58–67 %); however, the HpScl group had a significantly lower APOE ε4 allele frequency than all other groups (33 %, p < 0.05). We also examined the progranulin (GRN) rs5848 variant, where the T allele is associated with lower GRN expression and is reported to be a risk factor for frontotemporal lobar degeneration (FTLD), HpScl, and AD [9, 21, 30, 36]. Both the HpScl and HpScl-AD groups appeared to have a higher proportion of the GRN T allele (62 and 70 %, respectively), but only the HpScl-AD group achieved significance over the typical AD and LP-AD groups (53 and 48 %, respectively; p < 0.002). Furthermore, 21 % of the HpScl were homozygous for the T risk allele, which is higher than the frequency reported in FTLD (16 %) [32]. In comparison, the TMEM106B rs1990622 variant, where the C allele regulates GRN expression [11], is reported to be protective for AD with HpScl [32]. The TMEM106B C allele was found significantly less often in the HpScl and HpScl-AD groups (48 % for both) than the typical AD and LP-AD groups (70 and 74 %, respectively). Of note, none of HpScl cases were homozygous for the C allele. These findings imply APOE ε4 allele status is the best genetic indicator for AD, regardless of AD subtype (i.e., typical AD vs. LP-AD) or HpScl comorbidity, and the absence of the TMEM106B C allele is the best genetic indicator for HpScl, regardless of AD comorbidity.
Clinical and cognitive assessments
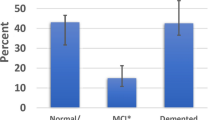
Table 3 summarizes all clinical findings. The age of disease onset was oldest in HpScl (79 years) and the youngest in typical AD (71 years). Disease duration was similar among AD, LP-AD, and HpScl (9–10 years), but was appreciably longer in HpScl-AD (12 years, p < 0.001) compared to AD and LP-AD. Family history of dementia was variably reported and ranged from 22 % in typical AD to 67 % in HpScl-AD. The percentage of cases given an amnestic clinical diagnosis was similar among LP-AD, HpScl-AD, and HpScl (88–93 %) but was somewhat lower in typical AD (78 %). To determine if cognitive tests could distinguish between study groups, we performed a retrospective analysis on a subset of cases with available MMSE scores. Longitudinal progression of cognitive decline was calculated for cases with at least three MMSE scores. Longitudinal decline was significantly slower in HpScl (−0.3 points/year) compared to all other groups (p ≤ 0.003), with typical AD exhibiting the steepest decline (−2.5 points/year, Fig. 3). These antemortem results show that HpScl patients are significantly older and present with a considerably slower cognitive decline than patients with AD, irrespective of AD subtype. To predict which of the significant clinical and genetic variables might improve the differential diagnosis of these late-life amnestic dementias, six logistic regression models were built (Supplementary Table 1). Detailed descriptions can be found in Supplementary results. Separately the presence versus absence of the GRN T allele and the presence versus absence of the TMEM106B C allele were investigated in HpScl and HpScl-AD for clinical and neuropathologic differences. No significant differences were found for age onset, disease duration, longitudinal decline on MMSE, family history, age at death, Braak NFT stage, or brain weight.

Longitudinal progression of cognitive decline in typical AD (a), LP-AD (b), HpScl-AD (c), and HpScl (d). A moving average (bolded black line) was plotted to illustrate the overall longitudinal decline in MMSE for each study group. AD Alzheimer’s disease, LP-AD limbic-predominant AD, HpScl hippocampal sclerosis
Discussion
By examining a large cohort of autopsy-confirmed cases with typical AD, LP-AD, HpScl-AD, and HpScl, we have identified substantial differences in clinical, genetic, and pathologic characteristics. The primary goal of this study was to directly compare HpScl and LP-AD because both diseases have focal medial temporal lobe atrophy and a similar age of onset, which can be difficult to distinguish. We also included typical AD as a comparison, as well as cases with concomitant AD and HpScl. Our results show that patients with HpScl tend to be very old at death, significantly older than typical AD. Furthermore, HpScl patients had a much slower rate of cognitive decline, which was significantly slower than the AD groups (AD, LP-AD, and HpScl-AD). Genetic comparison of our study groups demonstrated that MAPT H1 haplotype was not different between any of the study groups, except typical AD and LP-AD. However, all AD groups, including HpScl-AD, exhibited an over-representation of APOE ε4 carriers, while HpScl did not. In contrast, cases with HpScl and HpScl-AD had the increased risk allele in GRN and decreased protective allele in TMEM106B. Neuropathologic assessment revealed that HpScl tends to have less brain atrophy compared to the other groups, consistent with selective neuronal loss in medial temporal lobe. In keeping with the gestalt diagnosis, HpScl displayed minimal Alzheimer pathology compared to AD groups. The frequency of cerebrovascular disease pathology was highest in HpScl, but concomitant LBD pathology was rare. HpScl and HpScl-AD were selected for TDP-43 positivity [2, 24], which was most often Type A––confirming our earlier findings [2]. Typical AD and LP-AD, however, had a significantly lower frequency of TDP-43 pathology that is most often Type B [17, 23]. Although AD and HpScl are both late-onset dementias, it is apparent that AD and LP-AD share similar phenotypes that are often in sharp contrast to HpScl, whereas concomitant HpScl and AD exhibit features of both diseases. In distinguishing HpScl from LP-AD, we found significant differences in rate of decline on MMSE that will need to be followed up in a larger cohort. Encouragingly, both APOE and TMEM106B genotypes may be useful in the differential diagnosis.
The neuropathologic characteristics of typical AD and HpScl can be distinguished by brain weight, the regions affected, and the presence of pathologic proteins. Concomitant AD and HpScl, however, blur this distinction [4, 31]. HpScl-AD exhibits a similar NFT distribution and severity as typical AD in both the cortex and hippocampus. HpScl-AD also exhibits similar TDP-43 pattern and morphology as HpScl. Prior studies have demonstrated that 70–90 % of hippocampal sclerosis cases have TDP-43 lesions that are similar in morphology to those seen in FTLD [2, 24, 27]. We, and others, have also reported TDP-43 pathology in 23–56 % of AD cases [2, 3, 7]. Our current results suggest that the previously reported AD cohorts with high TDP-43 frequencies may have concomitant hippocampal sclerosis. Altogether, it appears that HpScl-AD brains are undergoing two distinctive neurodegenerative processes and, importantly, comorbidity of these diseases does not result in a synergistic effect [6, 15].
Prior studies have shown that HpScl and AD may share similar genetic risk factors as FTLD. Rademakers and colleagues [30] demonstrated that FTLD patients have an increased frequency of the homozygous T allele of GRN rs5848. This variant is located in a miRNA binding-site that regulates GRN expression. Homozygous T allele carriers have decreased GRN expression that is similar to FTLD patients with loss-of-function GRN mutations; therefore, this variant is considered a risk factor for FTLD. Subsequent reports have shown that the rs5848 variant is associated with hippocampal sclerosis in cases with a primary pathologic diagnosis of FTLD or AD [9, 29]. Here, we show for the first time that both HpScl-AD and HpScl without other significant pathology exhibit an increased frequency of the T allele. Surprisingly, 21 % of our HpScl are homozygous for the risk allele, which is higher than that observed in FTLD cases (16 %) [30]. Our typical AD group had a frequency similar to elderly controls that have been published [30]. A few contradictory reports have described an increased frequency of this genotype in clinical AD populations [21, 36]; however, these cases were not autopsy-confirmed and may include cases with HpScl. In contrast to the GRN rs5848 variant, the TMEM106B rs1990622 variant is protective against FTLD by regulating GRN levels or function [8, 11, 13, 35]. The protective CC genotype was only observed in 2.6 % of FTLD cases compared to 19 % of controls. A follow-up study showed that cases with both AD and HpScl had a decreased CC genotype frequency (6.8 %), but not AD alone [32]. Our current study corroborates those findings and additionally shows that none of our HpScl cases have the protective genotype. A recent genome-wide association study investigating hippocampal sclerosis as an endophenotype identified a novel polymorphism in the ATP-binding cassette, sub-family C member 9 (ABCC9) [26]. The study validated previous associations of GRN and TMEM106B as risk genes in hippocampal sclerosis, and did not find any association with APOE. Further genetic assessment of our cohort revealed APOE ε4, the strongest genetic risk factor for AD, is over-represented in the AD groups, including HpScl-AD. In contrast, APOE status was independent of risk for HpScl, which is consistent with previous reports [22, 27, 33]. In total, it appears that APOE status strongly influences AD neuropathology while GRN and TMEM106B genotypes strongly influence hippocampal sclerosis neuropathology. In the case of HpScl-AD, all of these genetic factors contribute to the mixed neuropathology. Furthermore, the HpScl-AD group exhibited the strongest family history of dementia that may be due to the combination of these genetic risk factors associated with two separate, yet similar, dementias.
Clinical detection of HpScl from other dementias like AD is needed for proper patient management, but this is a particularly difficult task. We determined that 93 % of HpScl were given a clinical amnestic diagnosis after a single neurologist (NRGR) reviewed available clinical notes. However, it should be noted that only 72 % of our HpScl were originally given an amnestic clinical diagnosis. Original non-amnestic diagnoses included dementia/mixed dementia (n = 3), frontotemporal dementia (n = 1), cerebrovascular accident (n = 1), and normal (n = 1). We have also demonstrated that slowly progressing longitudinal decline is a strong clinical indicator for HpScl. Others have reported that the cognitive decline in HpScl assessed by MMSE was similar to AD without HpScl, while AD with concomitant HpScl had a faster rate of decline than all other groups [25, 27]. These differing results may be due to different methodological approaches regarding number of tests or inclusion/exclusion of atypical AD cases. More detailed analyses are still needed to determine the significance of distinguishing HpScl using cognitive testing. Additionally, more work is needed to determine whether genotyping GRN and TMEM106B or measuring GRN in blood plasma may be useful in the differential diagnosis of HpScl with and without significant coexisting pathologies. Our neuropathologic findings lend strong evidence that topographic differences in tau, amyloid, and TDP-43 would be ideal targets for molecular imaging probes with positron emission tomography to visualize brain pathology. Of course, comprehensive assessments including cognitive testing, genetic profiling, and neuroimaging may provide the best ability to clinically discriminate late-onset amnestic diseases.
An inherent limitation of this study is that it is retrospective; therefore, only a subset of cases underwent the same clinical and cognitive assessments. Prospective longitudinal studies are needed to further define the clinical and neuropsychological characteristics of late-onset amnestic diseases. The major strength of this study includes a large sample size of autopsy-confirmed cases that were evaluated by a single neuropathologist for diagnostic and quantitative measures (i.e., TDP-43 typing and Thioflavin-S counts). Furthermore, neuropathologic classifications of AD subtypes were determined by an unbiased mathematical algorithm. These methods enabled us to determine that AD and HpScl are distinct amnestic diseases that occur in individuals of similar age.
References
Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, Hodges JR (2007) Focal cortical presentations of Alzheimer’s disease. Brain 130:2636–2645. doi:10.1093/brain/awm213
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. doi:10.1002/ana.21154
Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, Iritani S, Onaya M, Akiyama H (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. doi:10.1007/s00401-008-0480-1
Attems J, Jellinger KA (2006) Hippocampal sclerosis in Alzheimer disease and other dementias. Neurology 66:775. doi:10.1212/01.wnl.0000200959.50898.26 (pii):66/5/775
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Brenowitz WD, Monsell SE, Schmitt FA, Kukull WA, Nelson PT (2014) Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis 39:691–702. doi:10.3233/JAD-131880Q123N6P2Q0511V80
Davidson YS, Raby S, Foulds PG, Robinson A, Thompson JC, Sikkink S, Yusuf I, Amin H, DuPlessis D, Troakes C, Al-Sarraj S, Sloan C, Esiri MM, Prasher VP, Allsop D, Neary D, Pickering-Brown SM, Snowden JS, Mann DM (2011) TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol 122:703–713. doi:10.1007/s00401-011-0879-y
Deming Y, Cruchaga C (2014) TMEM106B: a strong FTLD disease modifier. Acta Neuropathol 127:419–422. doi:10.1007/s00401-014-1249-3
Dickson DW, Baker M, Rademakers R (2010) Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis 7:170–174. doi:10.1159/000289231
Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA (1994) Hippocampal sclerosis: a common pathological feature of dementia in very old (>or = 80 years of age) humans. Acta Neuropathol 88:212–221
Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, Forloni G, Kertesz A, Knopman DS, Uitti R, White CL 3rd, Caselli R, Lippa C, Bigio EH, Wszolek ZK, Binetti G, Mackenzie IR, Miller BL, Boeve BF, Younkin SG, Dickson DW, Petersen RC, Graff-Radford NR, Geschwind DH, Rademakers R (2011) TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76:467–474. doi:10.1212/WNL.0b013e31820a0e3b
Fotuhi M, Do D, Jack C (2012) Modifiable factors that alter the size of the hippocampus with ageing. Nat Rev Neurol 8:189–202. doi:10.1038/nrneurol.2012.27
Gallagher M, Suh E, Grossman M, Elman L, McCluskey L, Swieten J, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B, Rohrer J, Halliday G, Broeckhoven C, Seilhean D, Shaw P, Frosch M, Alafuzoff I, Antonell A, Bogdanovic N, Brooks W, Cairns N, Cooper-Knock J, Cotman C, Cras P, Cruts M, Deyn P, DeCarli C, Dobson-Stone C, Engelborghs S, Fox N, Galasko D, Gearing M, Gijselinck I, Grafman J, Hartikainen P, Hatanpaa K, Highley JR, Hodges J, Hulette C, Ince P, Jin L-W, Kirby J, Kofler J, Kril J, Kwok JJ, Levey A, Lieberman A, Llado A, Martin J–J, Masliah E, McDermott C, McKee A, McLean C, Mead S, Miller C, Miller J, Munoz D, Murrell J, Paulson H, Piguet O, Rossor M, Sanchez-Valle R, Sano M, Schneider J, Silbert L, Spina S, Zee J, Langenhove T, Warren J, Wharton S, White Iii C, Woltjer R, Trojanowski J, Lee VY, Deerlin V, Chen-Plotkin A (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127:407–418. doi:10.1007/s00401-013-1239-x
Galton CJ, Patterson K, Xuereb JH, Hodges JR (2000) Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 123(Pt 3):484–498
Graff-Radford NR, Murray ME, Ross OA, Duara R, Rademakers R, Whitwell JL, Dickson DW (2012) Differentiating clinicopathologic and genetic aspects of hippocampal sclerosis in Alzheimer’s disease from limbic predominant Alzheimer’s disease and “pure” hippocampal sclerosis. Alzheimer’s Dement 8:P620–P621
Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56:1095–1097
Janocko NJ, Brodersen KA, Soto-Ortolaza AI, Ross OA, Liesinger AM, Duara R, Graff-Radford NR, Dickson DW, Murray ME (2012) Neuropathologically defined subtypes of Alzheimer’s disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 124:681–692. doi:10.1007/s00401-012-1044-y
Jellinger KA, Attems J (2003) Incidence of cerebrovascular lesions in Alzheimer’s disease: a postmortem study. Acta Neuropathol 105:14–17. doi:10.1007/s00401-002-0634-5
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Jack CR, Jr, Parisi JE, Petersen RC, Dickson DW (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. doi:10.1007/s00401-014-1269-z
Khachaturian ZS (1985) Diagnosis of Alzheimer’s disease. Arch Neurol 42:1097–1105
Lee MJ, Chen TF, Cheng TW, Chiu MJ (2011) rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener Dis 8:216–220. doi:10.1159/000322538
Leverenz JB, Agustin CM, Tsuang D, Peskind ER, Edland SD, Nochlin D, DiGiacomo L, Bowen JD, McCormick WC, Teri L, Raskind MA, Kukull WA, Larson EB (2002) Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol 59:1099–1106 noc10247
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW (2011) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 10:785–796. doi:10.1016/S1474-4422(11)70156-9
Nelson P, Smith C, Abner E, Wilfred B, Wang W-X, Neltner J, Baker M, Fardo D, Kryscio R, Scheff S, Jicha G, Jellinger K, Eldik L, Schmitt F (2013) Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol 126:161–177. doi:10.1007/s00401-013-1154-1
Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR (2010) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20:66–79. doi:10.1111/j.1750-3639.2008.00244.xBPA244
Nelson PT, Estus S, Abner EL, Parikh I, Malik M, Neltner JH, Ighodaro E, Wang WX, Wilfred BR, Wang LS, Kukull WA, Nandakumar K, Farman ML, Poon WW, Corrada MM, Kawas CH, Cribbs DH, Bennett DA, Schneider JA, Larson EB, Crane PK, Valladares O, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Scheff SW, Sonnen JA, Haines JL, Pericak-Vance MA, Mayeux R, Farrer LA, Van Eldik LJ, Horbinski C, Green RC, Gearing M, Poon LW, Kramer PL, Woltjer RL, Montine TJ, Partch AB, Rajic AJ, Richmire K, Monsell SE, Schellenberg GD, Fardo DW (2014) ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. doi:10.1007/s00401-014-1282-2
Nelson PT, Schmitt FA, Lin Y, Abner EL, Jicha GA, Patel E, Thomason PC, Neltner JH, Smith CD, Santacruz KS, Sonnen JA, Poon LW, Gearing M, Green RC, Woodard JL, Van Eldik LJ, Kryscio RJ (2011) Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 134:1506–1518. doi:10.1093/brain/awr053
Neltner JH, Abner EL, Baker S, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Hammack E, Kukull WA, Brenowitz WD, Van Eldik LJ, Nelson PT (2014) Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 137:255–267. doi:10.1093/brain/awt318
Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR (2011) Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord 25:364–368. doi:10.1097/WAD.0b013e31820f8f50
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW (2008) Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet 17:3631–3642. doi:10.1093/hmg/ddn257
Rauramaa T, Pikkarainen M, Englund E, Ince PG, Jellinger K, Paetau A, Alafuzoff I (2013) Consensus recommendations on pathologic changes in the hippocampus: a postmortem multicenter inter-rater study. J Neuropathol Exp Neurol 72(452–461):4. doi:10.1097/NEN.1090b1013e318292492a
Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, Parisi JE, Petersen RC, Graff-Radford NR, Younkin SG, Dickson DW, Rademakers R (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79:717–718. doi:10.1212/WNL.0b013e318264e3ac
Troncoso JC, Kawas CH, Chang CK, Folstein MF, Hedreen JC (1996) Lack of association of the apoE4 allele with hippocampal sclerosis dementia. Neurosci Lett 204:138–140
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697. doi:10.1097/01.jnen.0000225908.90052.07
van Blitterswijk M, Mullen B, Nicholson A, Bieniek K, Heckman M, Baker M, DeJesus-Hernandez M, Finch N, Brown P, Murray M, Hsiung G-Y, Stewart H, Karydas A, Finger E, Kertesz A, Bigio E, Weintraub S, Mesulam M, Hatanpaa K, White Iii C, Strong M, Beach T, Wszolek Z, Lippa C, Caselli R, Petrucelli L, Josephs K, Parisi J, Knopman D, Petersen R, Mackenzie I, Seeley W, Grinberg L, Miller B, Boylan K, Graff-Radford N, Boeve B, Dickson D, Rademakers R (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 127:397–406. doi:10.1007/s00401-013-1240-4
Viswanathan J, Makinen P, Helisalmi S, Haapasalo A, Soininen H, Hiltunen M (2009) An association study between granulin gene polymorphisms and Alzheimer’s disease in Finnish population. Am J Med Genet B Neuropsychiatr Genet 150B:747–750. doi:10.1002/ajmg.b.30889
Acknowledgments
We thank the patients and their families who donated brains to help further our knowledge in neurodegeneration. The authors would like to acknowledge Linda G. Rousseau, Virginia R. Phillips, and Monica Castanedes-Casey for tissue processing and staining. The project was supported by the Einstein Aging Study (P01 AG03949), Mayo ADRC Grant (P50 AG16574), and the State of Florida Alzheimer’s Disease Initiative. MEM and this project were supported by the Robert and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program fellowship and the Mayo Clinic ADRC Pilot grant. DWD was supported by the Robert E Jacoby Professorship for Alzheimer’s Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. E. Murray and A. Cannon contributed equally to the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Murray, M.E., Cannon, A., Graff-Radford, N.R. et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 128, 411–421 (2014). https://doi.org/10.1007/s00401-014-1302-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1302-2