
Abstract
Tumors with histological features of pilocytic astrocytoma (PA), but with increased mitotic activity and additional high-grade features (particularly microvascular proliferation and palisading necrosis) have often been designated anaplastic pilocytic astrocytomas. The status of these tumors as a separate entity has not yet been conclusively demonstrated and molecular features have only been partially characterized. We performed DNA methylation profiling of 102 histologically defined anaplastic pilocytic astrocytomas. T-distributed stochastic neighbor-embedding (t-SNE) and hierarchical clustering analysis of these 102 cases against 158 reference cases from 12 glioma reference classes revealed that a subset of 83 of these tumors share a common DNA methylation profile that is distinct from the reference classes. These 83 tumors were thus denominated DNA methylation class anaplastic astrocytoma with piloid features (MC AAP). The 19 remaining tumors were distributed amongst the reference classes, with additional testing confirming the molecular diagnosis in most cases. Median age of patients with MC AAP was 41.5 years. The most frequent localization was the posterior fossa (74%). Deletions of CDKN2A/B (66/83, 80%), MAPK pathway gene alterations (49/65, 75%, most frequently affecting NF1, followed by BRAF and FGFR1) and mutations of ATRX or loss of ATRX expression (33/74, 45%) were the most common molecular alterations. All tumors were IDH1/2 wildtype. The MGMT promoter was methylated in 38/83 tumors (45%). Outcome analysis confirmed an unfavorable clinical course in comparison to PA, but better than IDH wildtype glioblastoma. In conclusion, we show that a subset of histologically defined anaplastic pilocytic astrocytomas forms a separate DNA methylation cluster, harbors recurrent alterations in MAPK pathway genes in combination with alterations of CDKN2A/B and ATRX, affects patients who are on average older than those diagnosed with PA and has an intermediate clinical outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
PA accounts for approximately 5% of gliomas across all age groups and for approximately 25% of all brain tumors encountered in pediatric neurosurgical practice [9, 51], thereby representing the most common primary brain tumor in patients of 0–19 years of age. In contrast, the majority of gliomas in adults are either diffuse gliomas with IDH1 mutation (either 1p19q co-deleted or 1p19q intact) or highly malignant glioblastomas (GBMs) [55]. Where gross total resection is feasible, clinical outcome of PA is usually favorable with 10-year overall survival rates of around 95% [9, 10, 14, 45]. The vast majority of PAs harbor single genetic alterations in genes encoding proteins of the mitogen-activated protein kinase (MAPK) pathway, with the most frequent findings being fusions of BRAF with KIAA1549 or rarely other genes, BRAF V600E point mutation, or alterations of NF1, FGFR1 or NTRK family genes [25]. Different localizations of PA are associated with different frequencies of molecular alterations, with posterior fossa tumors harboring a BRAF fusion in up to 80–90% of cases and supratentorial lesions showing this alteration in only about 60% of cases [21, 22]. Of further interest is the reported age association of BRAF fusions that seem to be less frequent in the adult population (e.g. 30% of patients of 31–40 years and 7% of patients older than 40 years of age) [17, 22]. To date, BRAF fusions are considered a molecular hallmark of WHO grade I PA with a typically favorable outcome [18, 21, 47]. Rare cases of PA, particularly in older patients, can have a more aggressive clinical behavior [3, 5, 44, 45, 51, 58]. The tumors of these patients may show anaplastic histological features including increased nuclear atypia, increased mitotic activity, prominent endothelial proliferation and/or palisading necrosis and have been discussed to represent a separate entity or subentity [45]. In the WHO classification 2016, this glioma subgroup is designated pilocytic astrocytoma with anaplasia. The identification of this particular subset of more aggressively behaving tumors is challenging, as the histological features described in the WHO classification do not exclude the diagnosis of conventional PA WHO grade I [9, 45], while histological overlap with pleomorphic xanthoastrocytoma (PXA)/anaplastic pleomorphic xanthoastrocytoma and GBM is also evident. Moreover, according to the WHO classification, grading and nomenclature of pilocytic astrocytoma with anaplasia are still to be conclusively defined [30]. One study on the molecular characterization of clinically aggressive/recurrent or histologically defined anaplastic pilocytic astrocytomas revealed a heterozygous PTEN/10q loss in 6 of 19 (32%) and a homozygous CDKN2A/B deletion in 3 of 15 (20%) cases with BRAF fusions identified in 63% of cerebellar lesions [44]. A different study observed an association with neurofibromatosis type I in 28% of histologically defined anaplastic pilocytic astrocytomas [16]. Nevertheless, more comprehensive approaches are necessary to establish the characteristic molecular profile of this tumor class.
Recently, DNA methylation profiling has become an important adjunct tool for tumor classification and identification of molecular subclasses [19, 32, 35, 36, 46, 49, 52, 53]. To molecularly characterize histologically defined anaplastic pilocytic astrocytomas, we analyzed a retrospective series of 102 cases with histological features of this putative entity by DNA methylation profiling, DNA copy number analysis, next generation gene panel sequencing as well as by histological characterization and correlated these data with clinical information. Herein, we report that a major subset (83/102, 81%) of these tumors show a common DNA methylation profile, harbor frequent MAPK pathway gene alterations and have additional molecular and clinical features that clearly distinguish them from WHO grade I PAs as well as from other adult diffuse gliomas.
Materials and methods
Tissue samples
102 cases which received the institutional or differential diagnosis of histologically defined anaplastic pilocytic astrocytoma between 1990 and 2016 were included in the study. The original diagnosis was taken for case selection for the series, so some selection bias due to changes in diagnostic criteria over the years cannot be excluded. Formalin-fixed and paraffin-embedded (FFPE) tissue of these cases was retrieved from the archives of the following neuropathology departments: Heidelberg, Mannheim, Charité Berlin, Bonn, Duesseldorf, Essen, Freiburg, Giessen, Hannover, Cologne, Leipzig, Muenster, Tuebingen, Wuerzburg, Zurich and from the following institutions: the Institute of Pathology, University of Bern; the Division of Neuropathology of the Johns Hopkins School of Medicine, Baltimore, Maryland, USA; the Mayo Clinic, Rochester, Minnesota, USA; the Division of Neuropathology, UCL Institute of Neurology in London; the Department of Cellular Pathology, Queen’s Hospital, Romford. In addition, we obtained material via the German Glioma Network. Tissue collection and processing as well as data collection were performed in compliance with local ethics regulations and approval.
Clinical data
The following clinical data were acquired, when possible: institutional diagnosis, patient gender, age at current diagnosis of histologically defined anaplastic pilocytic astrocytoma, localization, disease stage (primary surgery or re-resection), history of a precursor lesion, history of irradiation, presence of a tumor predisposition syndrome, time to re-surgery and overall survival. Time to recurrence was not uniformly available due to different definitions of recurrence and different modes of follow-up implemented in the various contributing centers. Criteria for outcome analyses are defined below.
Histological examination
Hematoxylin- and eosin (HE)-stained slides of 74/83 tissue samples of the MC AAP as well as all 19 tissue samples of the re-classified tumors were systematically reviewed for morphological criteria: general morphological growth pattern, cellularity, nuclear pleomorphism, mitoses, necrosis, vascular proliferation, eosinophilic granular bodies or Rosenthal fibers and infiltration pattern. The purpose of histological examination was to establish, as far as possible, histological features typical for the MC AAP.
Immunohistochemistry
For cases with available tissue, immunohistochemistry with antibodies specific for ATRX (n = 54), H3 K27M (n = 47), IDH1 R132H (n = 45) or BRAF V600E (n = 51) was performed on a Ventana BenchMark XT Immunostainer (Ventana Medical Systems, Tucson, Arizona, USA) using established protocols. For dilutions and antibody details, see Online Resource 1.
BRAF V600E, IDH1 R132H and H3 K27M immunohistochemistry was scored as either positive or negative. For all three antibodies, nonspecific staining of macrophages, eosinophilic granular bodies and calcified deposits was excluded from analysis. Staining of vessels or reactive glia was also not considered. Loss of nuclear ATRX expression was scored as specific, if over 80% of tumor cell nuclei showed loss of expression, while nuclei of non-neoplastic cells, such as endothelia, microglia, lymphocytes and reactive astrocytes, were positive. Of note, weak to moderate staining of tumor cell cytoplasm was occasionally seen and was considered as nonspecific [43]. Slides were scanned on a NanoZoomer Digital Slide Scanner (Hamamatsu, Japan) and photographed using Aperio ImageScope software (v11.0.2.725, Aperio Technologies, Vista, California, USA).
DNA extraction and quantification
DNA was extracted from FFPE tissue using the automated Maxwell system (Promega, Fitchburg, Massachusetts, USA) according to the manufacturer’s instructions. DNA concentration was determined using the Qubit dsDNA BR Assay kit (Invitrogen, Carlsbad, California, USA) following the producer’s guidelines.
Generation of DNA methylation array data and copy number profile calculation
From each tissue sample, 200–500 ng of DNA were processed for DNA methylation analysis. The Infinium HumanMethylation450 Bead-Chip (450k) array (Illumina, Carlsbad, California, USA) was used to determine the DNA methylation status of 482,421 CpG sites according to the manufacturer’s instructions at the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ). A copy number profile (CNP) was calculated from the methylation array data as previously described [20] using the ‘conumee’ package in R (http://bioconductor.org/packages/release/bioc/html/conumee.html). Amplifications in DNA copy number profile were defined as focal regions of copy number gain with a notably higher amplitude than regions of suspected single-copy gains.
H3F3A, BRAF, IDH1, IDH2 and TERT promoter mutation analysis by Sanger sequencing
Primer design and sequencing were performed according to standard protocols. Primer sequences and Refseq NM accession numbers for the respective genes are listed in Online Resource 2. For the amplification reaction, the following reagents were used: 12.5 µl of Go Taq G2 DNA polymerase (Promega), 1.25 µl of forward primer (10 pmol/µl), 1.25 µl of reverse primer (10 pmol/µl), 8.0 µl of nuclease-free water and 2 µl of template DNA (approximately 25 ng/µl). PCR conditions are listed in Online Resource 3.
Assessment of MGMT promoter methylation status
MGMT promoter methylation status was calculated from the methylation data as described by Bady et al. [4] with modifications: for each probe, an individual confidence interval for MGMT promoter methylation was calculated. If the calculated confidence interval included the cutoff value of 0.358 determined by Bady et al., MGMT promoter methylation status was defined as not determinable. For cases with an MGMT promoter methylation status not determinable by 450k methylation analysis, additional pyrosequencing was performed using the therascreen® MGMT Pyro® kit (QIAGEN®) and the PyroMark® Q24 system (QIAGEN®) according to the manufacturer’s protocol. Bisulfite conversion was done with the EpiTect fast DNA bisulfite kit (QIAGEN®). According to studies published by Quillien et al., Felsberg et al. and Reifenberger et al. [12, 39, 41], the cutoff value for MGMT promoter methylation status was set as follows: a mean MGMT promoter methylation percentage < 8% across the investigated CpG sites was considered as non-methylated and a value ≥ 8% was considered as methylated.
Gene panel sequencing
Gene panel sequencing was performed and resulting data were analyzed as previously described [48]. In brief, extracted DNA was sheared on a M220 Focused-ultrasonicator™ (Covaris®, Woburn, Massachusetts, USA). DNA integrity and fragment size were determined by the Bioanalyzer 2100 (Agilent, Santa Clara, California, USA). Sequencing was performed on a NextSeq 500 instrument (Illumina, Carlsbad, USA) with a mean coverage of 645 reads (with a standard deviation of 455 reads). Gene panel sequencing data were automatically annotated using annovar software that integrates information from databases such as dbSNP, the 1000 Genomes Project and COSMIC, as well as with SIFT and PolyPhen2 scores to infer the possible biological relevance of an alteration [56]. Gene panel sequencing data were then filtered applying the following criteria: first, exonic and splicing variants were selected. Among these, synonymous and stoploss variants were not further considered. Thereafter, variants with a frequency not exceeding 1% in the healthy population as well as undescribed variants were selected according to the 1000 Genomes Project database. Variants described as known polymorphisms in the single nucleotide polymorphism database (dbSNP, version 138: https://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?view+summary=view+summary&build_id=138) were not considered. Insertions and deletions were filtered for exonic frameshift changes that were not yet detected in the healthy population according to the 1000 Genomes Project database and that were not present in the single nucleotide polymorphism database (dbSNP). The remaining items (nonsynonymous, stopgain or splice-site variants, frameshift insertions and frameshift deletions) were evaluated for their potential clinic-pathological relevance using the COSMIC database (http://cancer.sanger.ac.uk/cosmic) and the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/). According to these databases, variants were categorized with regard to the probable consequences on protein function as follows: damaging, possibly damaging, likely not relevant or no applicable information. Variants assessed as damaging or possibly damaging were considered as mutations. Variants assessed as likely not relevant or with lack of information about clinical relevance were not considered. For the assessment of mutations in genes of the MAPK pathway, we considered NF1, BRAF, FGFR1, KRAS, NRAS, HRAS, NTRK2 and PTPN11. A full list of the genes represented in the applied gene panel is provided in Online Resource 4. The deFuse software tool (https://sourceforge.net/projects/defuse/, [33]) was used to identify KIAA1549:BRAF, FAM131B:BRAF, FGFR1:TACC1 and FGFR3:TACC3 fusions and to screen for NTRK2 fusions (QKI:NTRK2, NACC2:NTRK2). Other gene fusions—among these other BRAF fusions with rare fusion partners (RNF130:BRAF, CLCN6:BRAF, MKRN1:BRAF, GNAI1:BRAF)—were not sufficiently covered by this approach and, hence, were not identifiable by the applied methods.
Reference datasets
Reference datasets for t-SNE, clustering and copy number analyses
For t-SNE and hierarchical clustering, the following glioma reference classes were included: diffuse midline glioma, H3 K27 mutant (DMG K27; 14 cases); high-grade neuroepithelial tumor, H3 G34 mutant (GBM G34; 11 cases); GBM of the midline (GBM MID; 10 cases); GBM, mesenchymal subtype (GBM MES; 15 cases); GBM, RTK I subtype (GBM RTK I; 15 cases); GBM, RTK II subtype (GBM RTK II; 8 cases); pleomorphic xanthoastrocytoma (PXA; 17 cases); PA of the supratentorial hemispheres (PA SUP; 12 cases); PA of the posterior fossa (PA PF; 15 cases); PA of the midline (PA MID; 18 cases); dysembryoplastic neuroepithelial tumor (DNET; 13 cases) and diffuse leptomeningeal glioneuronal tumor (DLGNT; 10 cases).
Detailed descriptions of the reference methylation classes used in this study are outlined under https://www.molecularneuropathology.org [6, 7].
Reference sets for the Kaplan–Meier analysis
Outcome data were available for 41/83 of the MC AAP patients. For patients with an initial diagnosis of a lower-grade glioma, the date of the first diagnosis of histologically defined anaplastic pilocytic astrocytoma was considered as starting point for the outcome analysis. Kaplan–Meier analysis was performed in comparison to survival data from additional 281 glioma patients of 5 reference classes: 82 PAs WHO grade I, 18 PXAs, 26 anaplastic astrocytomas, IDH mutant (AIII IDH mut), 90 GBMs, IDH mutant (GBM IDH mut) and 56 GBMs, IDH wildtype (GBM IDH wt).
Statistical analyses
The DNA methylation array data were processed with the R/Bioconductor package minfi (version 1.20) [2]. For unsupervised hierarchical clustering of histologically defined anaplastic pilocytic astrocytomas and reference samples, we selected the 20,000 most variably methylated CpG sites across the dataset according to median absolute deviation. Pairwise similarity of samples was calculated using Euclidean distance. Clusters were then linked according to the Ward’s linkage method. The t-SNE plot was computed via the R package Rtsne [29] using the 20,000 most variable CpG sites according to standard deviation, 2000 iterations and a perplexity value of 20. To define the MC AAP, cutoffs for the X and the Y values were deduced from the t-SNE plot (Fig. 1) as follows: − 10 < Y < 10 and − 20 < X < 2. Survival data of MC AAP and reference group patients were evaluated via Kaplan–Meier analysis using the JMP software (SAS Institute, Cary, North Carolina, USA). Outcome analyses were calculated from the time point of the first histological diagnosis of anaplastic pilocytic astrocytoma. Observation time for the Kaplan–Meier analysis was cut at 10 years (120 months) for all subgroup analyses due to very small numbers of patients remaining under observation beyond this. To verify differences of the survival distributions between the respective patient groups, the log-rank test was applied.

t-SNE analysis of 102 cases with histological features compatible with the diagnosis of anaplastic pilocytic astrocytoma, indicated in black, and 158 reference cases of established glioma methylation classes, indicated in different colors. Tumors of the same class are depicted in one color. For this analysis, the 20,000 most variably methylated CPG sites were used. 83 of the histologically defined anaplastic pilocytic astrocytomas (colored in black) formed one group intermixed by only few other cases and were designated as DNA methylation class anaplastic astrocytoma with piloid features (MC AAP). 19 histologically defined anaplastic pilocytic astrocytomas showed similarities to reference methylation classes, and therefore, grouped within these classes in the t-SNE. Reference methylation classes: GBM G34 glioblastoma, IDH wildtype, H3 G34 mutant; DMG K27 diffuse midline glioma, H3 K27M mutant; GBM MID glioblastoma, IDH wildtype, subclass midline; GBM MES glioblastoma, IDH wildtype, subclass mesenchymal; GBM RTK I glioblastoma, IDH wildtype, subclass RTK I; GBM RTK II glioblastoma, IDH wildtype, subclass RTK II; PXA (anaplastic) pleomorphic xanthoastrocytoma; DLGNT diffuse leptomeningeal glioneuronal tumor; PA SUP low-grade glioma, subclass hemispheric pilocytic astrocytoma and ganglioglioma; PA MID low-grade glioma, subclass midline pilocytic astrocytoma; PA PF low-grade glioma, subclass posterior fossa pilocytic astrocytoma; DNET low-grade glioma, dysembryoplastic neuroepithelial tumor
Results
A subset of histologically defined anaplastic pilocytic astrocytomas share a common DNA methylation profile
We performed DNA methylation analysis of 102 gliomas with the institutional or differential diagnosis of anaplastic pilocytic astrocytoma. Unsupervised t-SNE analysis of their DNA methylation profiles against 158 reference cases from 12 different glioma DNA methylation classes revealed a DNA methylation cluster comprising 83 of these 102 tumors (81%; Fig. 1). Subgroups within the main group of 83 tumors did not become apparent in the t-SNE analysis. The 19 remaining tumors grouped to (or close to) one of the reference classes. Two of these showed high similarity to high-grade glioma reference classes (one to DMG K27 and the other to GBM MID), two to the PXA reference class and 15 cases were similar to the low-grade glioma reference classes comprising PAs and DNETs. The 83 tumors in the cohort of histologically defined anaplastic pilocytic astrocytomas, henceforth denominated DNA methylation class anaplastic astrocytoma with piloid features (MC AAP), were further characterized by integration of clinical and molecular features. Of note, the MC AAP also contained two tumors with a H3 K27M mutation. Indeed, these lesions were located near the midline (one in the third and lateral ventricle and the other in the cerebellum), but did not otherwise show histological features of DMG K27, as both cases had a piloid morphology. Notably, the MC AAP showed similarity with the methylation profiles of the DLGNT reference class. Furthermore, two PA MID reference cases clustered to the MC AAP.
In addition, we performed an unsupervised clustering analysis of the same cohort as analyzed by t-SNE (Online Resource 5). This analysis revealed a cluster in which 81 tumors were congruent with the 83 MC AAP tumors identified by t-SNE, confirming the assignment as a separate DNA methylation cluster. Within this methylation cluster, two subclusters A and B (Online Resource 5) were putatively identified. There was, however, no high consistency in repeated clustering analyses and no obvious clinical, histological or molecular differences to further suggest treating these as two independent subclusters.
Re-classification of tumors with histologically defined anaplastic pilocytic astrocytomas, but divergent DNA methylation profile
t-SNE and clustering analysis revealed that 19 of the 102 tumors with histological features of anaplastic pilocytic astrocytoma displayed a DNA methylation profile that differed from the MC AAP (Fig. 1 and Online Resource 5). In keeping with our experience with other series [7, 53], the majority of these tumors could be re-classified on the basis of re-evaluation of histology and orthogonal molecular findings (immunohistochemistry for BRAF V600E, H3 K27M, copy number profile analysis): six tumors were downgraded to PA WHO grade I, one case was re-classified as PXA, one as diffuse midline glioma with H3 K27M mutation, one as GBM, IDH wildtype and two as low-grade gliomas, not elsewhere classified (NEC). The remaining eight cases could not be definitely classified. Online Resources 6, 7 and 8 give details and show examples of re-classified and non-classifiable cases.
Clinical characteristics of the MC AAP
The tumors in the MC AAP (n = 83) were preferentially located in the posterior fossa (74%), most frequently in the cerebellum (63%). 17% were located supratentorially, two cases (2%) had a supra- and infratentorial localization and five cases (7%) were located in the spinal compartment (Fig. 2a). Median patient age at resection was 41.5 years with only 8 out of 76 tumors (11%) occurring in patients below the age of 20 years (Fig. 2b). The distribution of male and female patients was balanced (male:female ratio of 1.1; n = 83). Of 67 patients with available data, 42 patients (63%) presented with primary surgery and without previously known lesions, whereas the investigated tumors of 25 patients (37%) originated from a re-resection (Fig. 2c). For 8 of these cases (12%) a low-grade primary tumor was known, 3 cases (4%) were initially diagnosed with histologically defined anaplastic pilocytic astrocytoma and 14 cases (17%) had primary tumors with unknown initial grade. Of the eight patients with a reported low-grade precursor tumor, six had been historically diagnosed as PA, one as oligodendroglioma and one as diffuse astrocytoma. Unfortunately, material of these cases was not available for histological or molecular re-evaluation. For the 25 re-resected lesions, second surgery was performed within 10 years in 18 cases (72%), whereas time to re-surgery was longer than 10 years in four cases (16%) and was unknown for another three cases (12%). Median time to re-surgery was 1.2 years with a range from 66 days up to 28.3 years. For patients with a low-grade primary tumor, median time to re-surgery was 3.2 years (n = 6/8), whereas in patients with a high-grade primary tumor, median time to re-surgery was only 0.9 years (n = 3/3). For only 5% (4 cases) of the patients in the whole cohort, previous brain irradiation was documented. A diagnosis of neurofibromatosis type 1 was clinically known in one patient and clinically suspected in a second patient. We further cannot exclude that some of the other NF1 mutations identified in the tumor series (see below) may have also been present in the germline. Other hereditary tumor predisposition syndromes were not documented.

Clinical characteristics of the DNA methylation class AAP: a tumor localization throughout the central nervous system; b age distribution; c distribution of clinical presentation as either primary lesion or as lesion with either high grade, low-grade or unknown grade precursor lesion
Histological and immunohistochemical characteristics of MC AAP tumors
We were able to comprehensively evaluate 74/83 cases of the MC AAP histologically including the assessment of growth and infiltration pattern, cellularity, nuclear pleomorphism, mitotic count, presence of necrosis, vascular proliferation and presence of eosinophilic granular bodies and/or Rosenthal fibers. The remaining nine tumors were not comprehensively evaluable because of limited paraffin material. Results are summarized in Fig. 3. Examples for histological features of the MC AAP are shown in Fig. 4a–c. In our series, features of cortical dysplasia adjacent to the tumors were not obvious in any of the cases.

Histological features of the DNA methylation class AAP: in n = 74 cases sufficient material for evaluation was available

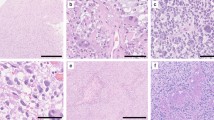
Histology and immunohistochemistry of DNA methylation class AAP (MC AAP): a HE stained section depicting a moderately cell dense, moderately pleomorphic glioma with multiple eosinophilic granular bodies (black arrows), a mitotic figure (white arrow in the inset) and a Rosenthal fiber (white asterisk in the inset; 400-fold magnification); b HE of another MC AAP tumor with glomeruloid vessel proliferation (asterisks; 400-fold magnification); c HE of another, more piloid MC AAP tumor with patchy necrosis in the lower left of the micrograph (400-fold magnification); d ATRX immunohistochemistry (400-fold magnification), the tumor cell nuclei show loss of ATRX expression, whereas nuclear ATRX expression in vessels and inflammatory cells is retained; e H3 K27M immunohistochemistry (400-fold magnification), negative H3 K27M staining in all but one case; f IDH1 R132H immunohistochemistry (400-fold magnification), all investigated lesions were negative for IDH1 R132H
Immunohistochemical analysis included assessment of the glioma markers ATRX, BRAF V600E, H3 K27M and IDH1 R132H. In 44% (24/54) of MC AAP lesions nuclear ATRX expression was lost in the tumor cells (Fig. 4d). BRAF immunohistochemistry revealed only one positive case (1/51, 2%). By H3 K27M immunohistochemistry and confirmatory Sanger sequencing analysis, one case (1/47, 2%) with a H3 K27M mutation was identified (Online Resource 9). For all the remaining cases tested by immunohistochemistry, H3 K27M (Fig. 4e) and BRAF V600E were negative. IDH1 R132H immunohistochemistry was negative in all cases investigated (45/45) (Fig. 4f).
DNA copy number aberrations in the MC AAP: CDKN2A/B deletion represents the most frequent structural aberration
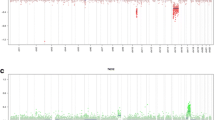
Copy number profile analysis of the MC AAP tumors disclosed numerous, partially complex chromosomal alterations: in 73 of 83 cases (88%), more than three structural aberrations were found, whereas a small subset of 10 cases (12%) exhibited three or fewer alterations. Figure 5a illustrates a representative copy number profile of a PA in the posterior fossa, whereas in Fig. 5b, an example of a copy number profile for a tumor of the MC AAP is shown. Figure 5c and d show a summary plot of chromosomal aberrations of 45 PA reference cases (comprising 15 cases of posterior fossa PA, 18 cases of midline PA and 12 cases of supratentorial PA) and from all 83 tumors of the MC AAP. The most frequent aberration in the MC AAP (66/83 cases, 80%) was a deletion of CDKN2A/B, which was frequently associated with broader deletions or complex changes on chromosome arm 9p (41/83 cases, 49%). A further significant finding was a BRAF fusion indicated by a focal low level peak indicative of a tandem duplication on chromosome 7q in 15 out of 74 assessed cases (20%) (Figs. 5b, d, 6). Other recurrent copy number aberrations were gains and/or complex changes on chromosome arms 12q (27%) and 17q (33%) as well as deletions on chromosomes 1p (19%), 8p (23%) and 19q (22%). A CDK4 amplification was detected in seven cases (8%) and this was associated with a concomitant MDM2 amplification in four cases (5%). Other recurrent amplifications more commonly seen in GBM (e.g., of MDM4, PDGFRα, EGFR, CDK6, MET) were not observed.

Single case examples and summary copy number profiles (CNPs) of conventional pilocytic astrocytoma (PA) and the methylation class anaplastic astrocytoma with piloid features (MC AAP): a example of a CNP of conventional PA, the blue arrow indicates a KIAA1549:BRAF fusion, other CNP aberrations are rare; b example of a CNP of an MC AAP tumor, the blue arrows indicate a KIAA1549:BRAF fusion and a CDKN2A/B deletion, numerous further chromosomal gains and losses are apparent; c summary CNP of conventional PA indicating a high fraction of cases with a KIAA1549:BRAF fusion (blue arrow) and whole chromosomal gains in rare cases, other chromosomal aberrations including chromosomal losses are rarely encountered; d summary CNP of MC AAP with a smaller proportion of cases harboring KIAA1549:BRAF and a high proportion of cases with CDKN2A/B deletion (both indicated by blue arrows), numerous further chromosomal gains and losses are observed, particularly gains of chromosome 12q and 17q, losses of chromosomes 1p, 8p and 19q as well as gains, losses or complex changes on chromosome 9p

Summary of molecular alterations and patient characteristics for the methylation class anaplastic astrocytoma with piloid features. For molecular alterations, black fields indicate the presence and light blue fields the absence of the respective alteration. Grey fields indicate that the lesion was not tested for the specific alteration or that the respective parameter was not determinable. mut mutation, del deletion, fus fusion, meth methylated
MC AAP tumors harbor mutations in MAPK pathway genes
Alterations affecting genes encoding members of the mitogen-activated protein kinase (MAPK) pathway have previously been found to occur in up to 100% of WHO grade I PAs [22–24]. By combining immunohistochemistry, panel sequencing and copy number analysis, we were able to comprehensively examine the status of MAPK pathway gene alterations for 65 of 83 tumors of the MC AAP (Fig. 6). In 49 out of these 65 cases (75%), at least one characteristic MAPK pathway gene alteration was detected. In notable contrast to typical PAs, the most frequently affected gene was NF1, being altered in 20 of 67 lesions (30%). In total, five tumors exhibited a deletion, ten cases harbored a mutation (including one clinically known NF1 syndrome patient) and three cases showed both, a deletion in the copy number profile and a mutation of the NF1 gene. The remaining two tumors showed immunohistochemical loss of NF1 protein expression and a balanced NF1 gene dosage in the copy number profile. Since matching constitutional DNA was unfortunately lacking for most cases, we cannot comment on the rate of germline versus somatic NF1 alterations in the series. BRAF was the second most frequently altered MAPK pathway gene: 15 of 74 cases (20%) harbored a BRAF fusion and one case (1%) a BRAF V600E mutation. In 12 out of 64 cases (19%), an FGFR1 alteration was detected. Among these, 11 of 64 tumors (17%) had a point mutation at one of the known hotspots (K656E/N, N546D/K) and one case (2%) exhibited an FGFR1:TACC1 fusion. In another two of 64 cases (3%) a KRAS point mutation (Q61H in one and V14A in the other) was detected (Fig. 6). MAPK pathway gene alterations were mutually exclusive with the exception of one case harboring both an NF1 and an FGFR1 alteration (Fig. 6). The deFuse software tool was used to identify KIAA1549:BRAF, FAM131B:BRAF and FGFR1:TACC1 fusions and to screen for NTRK2 fusions. Other gene fusions were not sufficiently covered by our gene panel and were thus not identifiable by our approach. RNA sequencing was not performed.
MC AAP tumors frequently display alterations of ATRX, but rarely of TERT
Alterations in mechanisms of telomere maintenance are well known drivers of tumor progression in a variety of entities. In particular, TERT promoter and ATRX mutations are frequently described in diffuse gliomas and have been established as useful markers for their classification and prognostication [38, 42, 43]. Unexpectedly, we found a high rate of ATRX alterations in the MC AAP. 74 cases were evaluable for ATRX alterations either by immunohistochemical stainings and/or by panel sequencing analysis. From these 74 tumors, 33 (45%) displayed either a loss of ATRX expression and/or an ATRX mutation. In 15 of 64 tumors, (23%) mutant ATRX was detected by panel sequencing and 24 of 54 tumors (44%) showed an immunohistochemical loss of nuclear ATRX expression. For 21 of the latter 24 cases panel sequencing data were also available: among these, an ATRX mutation was confirmed in 12 cases (57%). In the 18 cases with retained nuclear ATRX expression and with available panel sequencing data, no ATRX mutation was detected. In summary, an ATRX mutation was confirmed in a subset of tumors with immunohistochemical loss of ATRX expression by gene panel sequencing. Hence, the sensitivity of our gene panel sequencing approach to predict ATRX protein loss was 57%, the specificity being 100%. In a study on IDH mutant gliomas, ATRX gene alterations were also not found in a subset of cases with a loss of nuclear ATRX expression, suggesting alternative mechanisms of inactivation [59].
ATRX alterations and TERT promoter mutations are known to occur in a mutually exclusive manner in gliomas [26, 43]. To determine the rate of TERT promoter mutations in the MC AAP and to explore, whether these are mutually exclusive with ATRX alterations in this newly defined tumor class, we performed Sanger sequencing of the TERT promoter region for 74 tumors of the MC AAP from which 33 cases carried an ATRX alteration (Fig. 6). Two tumors (3%) with a TERT promoter mutation, both with the nucleotide exchange C228T, were identified. Both tumors did not harbor an ATRX alteration. While gain of chromosome 5p was observed in approximately 10% of MC AAP tumors, no focal TERT amplifications were seen (Fig. 5c).
MGMT promoter hypermethylation is a frequent finding in MC AAP tumors
In 38 out of 83 tumors of the MC AAP (46%), the MGMT promoter was hypermethylated. Another 38 tumors (46%) showed a non-methylated MGMT promoter and in seven tumors (8%), the MGMT promoter methylation status as assessed by array analysis was not determinable and MGMT promoter pyrosequencing could not be performed due to lack of material (Fig. 6).
Comparison of the MC AAP with the molecular subtypes of glioma previously described by Ceccarelli et al.
Ceccarelli et al. have recently identified a subset of adult diffuse IDH wildtype gliomas within The Cancer Genome Atlas (TCGA) data showing a DNA methylation profile distinct from the classic-like and mesenchymal-like GBM defined by Sturm et al. [8, 54]. This subset of tumors has been shown to be further subdivided into two DNA methylation subclasses, of which one shows similarity to GBM and the other to PA. These DNA methylation subclasses have been designated “LGm6-GBM” and “PA-like low-grade gliomas” (LGG) [8]. To explore the relatedness of DNA methylation profiles of the MC AAP (n = 83) to the PA-like LGG cohort of the TCGA series (n = 29 tumors of 26 patients), we performed a t-SNE analysis of the two series together with the established reference classes (Fig. 7). While 5 of 29 PA-like LGG clustered more closely to GBM and 14 of 29 clustered more closely to low-grade glioma reference classes, the DNA methylation profiles of 10 of these 29 TCGA tumors (seven patients) showed high similarity to the MC AAP. Interestingly, the tumors of these seven patients were also enriched for ATRX, but not for BRAF alterations [8]. Among the 5 TCGA tumors which fell into the GBM reference classes, one with a H3 G34 and one with a H3 K27 mutation was reported [8]. Indeed, these two cases clustered as expected to the GBM G34 and the DMG K27 reference methylation class, respectively.

t-SNE of DNA methylation profiles of 102 cases with histological features compatible with the diagnosis of anaplastic pilocytic astrocytoma (APA hist; blue) and 158 reference gliomas colored identically to Fig. 1 together with 29 “PA like low-grade gliomas” (PA-like LGG) from The Cancer Genome Atlas [9] (black triangles) using the 20,000 most variably methylated CPG islands. Ten of 29 PA-like LGG tumors (7 patients*) showed highest similarity to the MC AAP (falling in the same area within the t-SNE) while 14 tumors show higher similarities with low-grade glioma reference classes and 5 tumors show methylation profile similarities with glioblastoma classes. For group abbreviations see legend of Fig. 1. * Two patients are represented with primary tumor and recurrence(s): 1/1*/1**- primary tumor/first recurrence/second recurrence of patient 1; 2/2*- primary tumor/recurrence of patient 2
Outcome analyses
As this study is a retrospective analysis, availability of clinical data was limited. Outcome data were available for 41 of the 83 patients with an MC AAP tumor: by the date of last follow-up, 18 patients (44%) had died and 23 patients (56%) were alive. Kaplan–Meier analysis of these 41 patients against outcome data of five glioma reference classes showed a survival probability inferior to patients with conventional PA, PXA and IDH mutant anaplastic astrocytoma and comparable outcome to patients with IDH mutant GBM, but superior to patients with IDH wildtype GBM (Fig. 8a). Of note, univariate outcome analysis of patients of the MC AAP with a characteristic MAPK pathway gene alteration identified in this study compared to patients for whom such an alteration was not detected showed a significantly better prognosis (p = 0.032, Fig. 8b). Kaplan–Meier analysis for the detected individual MAPK pathway gene alterations alone was also performed and showed no significant differences of survival probabilities between patients with BRAF, NF1 or FGFR1 altered tumors and patients negative for a characteristic MAPK pathway gene alteration (Online Resource 10). No significant outcome differences were seen in patients with MGMT methylated versus non-methylated tumors (p = 0.922) as well as in patients with ATRX altered versus ATRX wildtype tumors (p = 0.685) (Fig. 8c, d). Furthermore, outcome analysis dependent on histological criteria (presence/absence of necrosis, mitotic count) was performed (n = 38). No significant difference in survival probability between tumors with versus without necrosis (p = 0.468) or with 0, 1–2 versus more than 2 mitoses (p = 0.383) was evident (Online Resource 11). Among the 38 cases with available survival data and comprehensive histological review, only two cases showed palisading necrosis. Therefore, separate testing for this feature in outcome analysis was not feasible. Kaplan–Meier analysis was also performed separately for patients with tumors diagnosed with histologically defined anaplastic pilocytic astrocytoma on first resection vs. those that were diagnosed with histologically defined anaplastic pilocytic astrocytoma on re-resection (Online Resource 12). Survival of these two groups did not differ significantly. Due to retrospective inquiry of clinical data for the patient cohort in this study, treatment information such as extent of surgery, application of chemotherapy or preoperative performance status were not documented in a systematic or standardized fashion. Therefore, a comprehensive data set concerning these factors could not be compiled.

Outcome analysis of patients with tumors of the methylation class anaplastic astrocytoma with piloid features (MC AAP): a Kaplan–Meier analysis of overall survival of MC AAP in comparison to reference glioma classes indicates an intermediate outcome reminiscent of IDH mutant glioblastoma: b MC AAP outcome stratified by detection of a characteristic MAPK pathway alteration in this study indicates a worse outcome when no such alteration is detected; c no associations with outcome were observed for MGMT promoter methylation status or for d stratification into cases with altered or retained ATRX. From 41 cases with survival data, information about the presence or absence of a MAPK alteration was available for 34 patients, information about methylation or non-methylation of the MGMT promoter was available for 39 patients and information about ATRX status was available for 38 patients. PA conventional pilocytic astrocytoma; PXA pleomorphic xanthoastrocytoma; A III IDH anaplastic astrocytoma, IDH mutant, WHO grade III; GBM IDH mut glioblastoma, IDH mutant, WHO grade IV; GBM IDH wt glioblastoma, IDH wildtype, WHO grade IV; mut mutation; alt alteration; charact. characteristic; meth methylated; unmeth unmethylated, wt wildtype; OS overall survival
Discussion
In this study, we report a novel class of glial tumors with a common DNA methylation profile including many cases with a morphological overlap with PA with anaplasia as described in the WHO classification 2016 [30]. However, detailed histological evaluation shows a wide range of morphological features that would occasionally be more in line with other tumor entities (especially high-grade glioma/GBM). Thus, as with a growing number of CNS tumors, the tumor class defined here by DNA methylation profiling (MC AAP) is not exactly congruent with PA with anaplasia as defined in the current WHO classification [30].
Initially, t-SNE and cluster analysis of the DNA methylation profiles were performed and consistently revealed a DNA methylation cluster denominated MC AAP (anaplastic astrocytoma with piloid features). The closest resemblance of the DNA methylation profiles of these tumors was evident with the reference DNA methylation class of DLGNT (Fig. 1, Online Resource 5). Indeed, these tumors may be somehow related to PA with anaplasia, since they also frequently harbor BRAF fusions, but additionally show other alterations not typically found in classical PA (e.g., 1p loss) [6]. In the t-SNE analysis, two cases with H3 K27M mutation also clustered to the MC AAP, indicating that this mutation may also occur in these tumors. This adds to the notion that H3 K27M mutations are not 100% specific for DMG K27 but may also occur in other tumors [60]. Further, two PA MID reference tumors clustered among the MC AAP. This may indicate a certain degree of closer relation of these tumors to the MC AAP, but may also be related to technical limitations of t-SNE analyses to classify individual tumors. Analysis of a higher number of cases is required to further explore the profiles of such histologically low-grade lesions clustering to the MC AAP and to identify possible subgroups within the MC AAP.
Our further investigations focused on the clinical, histological and molecular characterization of the MC AAP. As also previously reported for a series of histologically defined anaplastic pilocytic astrocytomas [45], we observed a higher median patient age in the MC AAP than in conventional PAs [5]. In fact, gliomas belonging to the MC AAP mostly arise in adults. Only 11% (8/76) of these tumors occurred in patients less than 20 years of age. The most frequent tumor localization in our series was the posterior fossa (74%) with 63% of the tumors originating in the cerebellum. Compared to a previously reported series of histologically defined anaplastic pilocytic astrocytomas [45], the posterior fossa localization seems even more frequent among gliomas of the MC AAP. Importantly, clinical history of radiotherapy was reported in only 5% (4/83) of the patients. Thus, the role of irradiation in progression from conventional PA to anaplasia (and indeed, whether such progression occurs at all as opposed to de novo occurrence of an anaplastic lesion) may require additional investigation [1, 45]. Another issue of interest was the exploration of differences in the molecular profiles between tumors which were primarily anaplastic and tumors reported to develop from lower-grade lesions (secondarily anaplastic). Differences between these two groups were not obvious in this study. We are currently compiling a series of classic PAs that later recurred as anaplastic tumors to explore if the initial tumor already harbored specific molecular features.
As shown in Fig. 3, we observed a wide spectrum of histological features in our series: most of the tumors in the MC AAP showed a piloid or GBM-like general morphological pattern, were moderately cellular, had moderate nuclear pleomorphism, at least one mitosis per ten HPF and microvascular proliferation. Around one-third of the tumors exhibited areas of necrosis and one-third showed eosinophilic granular bodies or Rosenthal fibers, respectively. In summary, morphological characteristics of the MC AAP were not particularly specific and may overlap with other low- and high-grade gliomas. Despite evidence of typical histological features of anaplasia in a small subset of tumors, our investigations revealed that the histology of the majority of cases in our series appears rather inconspicuous. Thus, in an analogous fashion to other tumors such as IDH mutant gliomas, molecular analysis may be required to come to a final, integrated diagnosis. With regard to immunohistochemistry, the most interesting finding was a loss of nuclear ATRX expression in a subset of MC AAP tumors. Besides, Ebrahimi et al. previously did not detect ATRX alterations in conventional PAs, but identified one case of histologically defined anaplastic pilocytic astrocytoma and one case of BRAF-fused cerebellar GBM with loss of ATRX expression [11]. Notably, an ATRX mutation was verified in only a major subset of MC AAP tumors. As in a previous study on IDH mutant gliomas, an ATRX mutation was not found in all cases with lost ATRX expression. It, therefore, appears likely that other mechanisms besides exonic mutations, such as small deletions, may also result in a functional (and immunohistochemical) loss of ATRX protein [59].
MGMT promoter methylation status has been shown to have predictive and prognostic value, particularly for patients with GBM [15, 27, 34]. This study revealed that MGMT promoter hypermethylation was present in approximately half of MC AAP tumors, whereas in a control cohort of conventional PAs (n = 45), the MGMT status was either non-methylated in most of the cases (35/45, 78%) or not determinable in a subset (10/45, 22%) of tumors. These data indicate that MGMT promoter methylation status may be of some diagnostic relevance in terms of distinguishing conventional PA from AAP. However, the present data do not provide any evidence for an association of MGMT promoter methylation status with OS amongst the MC AAP. Admittedly, the prognostic value of this outcome analysis is very limited, as clinical information about previous chemotherapy and applied pharmaceuticals (e.g., temozolomide) were not available for most of the cases.
Another important finding of this study was that the distribution of MAPK pathway gene alterations in MC AAP clearly differed from that known for conventional PA. In particular, BRAF fusions are known to be the most abundant molecular alteration in conventional PAs with a frequency of up to 75%, depending on tumor localization and patient age. The second most common alteration in approximately 7% of PAs involves the NF1 gene followed by activating BRAF mutations (5%) and FGFR1 hotspot mutations (5%). Rarely (about 1 up to 2% each) NTRK2 fusions, PTPN11 mutations and RAF1 fusions were described [22, 25]. In contrast, with a frequency of only 20%, BRAF fusions were surprisingly rare in the MC AAP, whereas NF1 turned out to be the most frequently affected gene being altered in 30% of the tumors. It was not possible to determine germline NF1 status in most cases. Furthermore, a substantial fraction (19%) of tumors harbored an FGFR1 alteration. Among these, in one tumor, an FGFR1:TACC1 fusion was detected, as previously reported in pediatric low-grade as well as in high-grade gliomas. This aberration has also been shown to transform primary astrocytes into highly proliferating glial tumors [50, 60]. NTRK2 fusions and PTPN11 mutations were not observed and a KRAS mutation was detectable in only two cases (3%). FGFR1 alterations have been described to be preferentially found in extra-cerebellar and especially in midline gliomas [9, 25]. However, in the present series, the majority of FGFR1 mutant cases were located in the posterior fossa (Fig. 6). Hence, these data indicate that FGFR1 alterations may not be restricted to supratentorial midline localizations. BRAF alterations have been described to be less frequent (36% of cases) in adult compared to juvenile PAs [37]. In a study conducted by Hasselblatt et al., comparable results were obtained with a fraction of BRAF fusions of 30% in patients aged 31–40 years and of 7% in patients older than 40 years [17]. As MC AAP is also characterized by a higher patient age, the age distribution of BRAF fusions may explain the comparably low fraction of tumors positive for a BRAF fusion in the present series. By analysis of copy number profiles of classical PAs and MC AAP tumors we observed that CDKN2A/B deletion in a BRAF fusion-positive astrocytic glioma is highly suggestive for the diagnosis of AAP and makes the diagnosis of WHO grade I PA highly unlikely.
Aberrant activation of the MAPK pathway in PA, particularly by BRAF or RAS activation and NF1 inactivation, is thought to trigger oncogene-induced senescence (OIS) via engagement of the cyclin-dependent kinase inhibitors p16Ink4a, the gene product of CDKN2A, and/or p21Waf1. As these proteins trigger cell cycle arrest, they are thought to be associated with the indolent biological and clinical behavior of conventional PAs [22]. Rodriguez et al. observed a loss of p16Ink4a in a subset of PAs with anaplastic features [44]. Another study revealed that PA patients with immunohistochemical loss of p16Ink4a expression had a shorter overall survival than PA patients with retained p16Ink4a expression [40]. These findings raised the question, whether the prognosis of patients with an MC AAP tumor may be associated with the presence or absence of p16Ink4a. Due to the low case number of only four patients with a balanced CDKN2A/B copy number and available survival data, outcome analysis for this parameter could not be performed. Approximately half of the tumors comprising the MC AAP displayed a MAPK pathway gene alteration in combination with a CDKN2A/B deletion. For these tumors, impairment of OIS may be likely. ATRX has recently been suggested to also play a role in oncogene-induced senescence: Kovatcheva et al. showed that ATRX binds to oncogene-induced senescence-associated gene loci, particularly to the HRAS gene locus, and represses their activity. Conversely, high expression of HRAS is supposed to prevent senescence induction [28]. Based on these data, it could be hypothesized, that acquisition of an ATRX alteration may represent another mechanism of impairing oncogene induced senescence in MAPK-altered tumors, and that this may contribute to the adverse biological and clinical behavior of AAPs.
Existence of anaplastic features in PA has previously been found to be associated with decreased overall survival. In one series, outcomes of patients with histologically defined anaplastic pilocytic astrocytomas were indeed less favorable in comparison to conventional PA patients. Survival data were stated as comparable to WHO grade II and III diffusely infiltrative astrocytomas, but still better than grade WHO IV GBMs [45]. Another study suggested that patients with histologically defined anaplastic pilocytic astrocytomas may have a better prognosis than other high-grade gliomas in adult patients [13]. However, Kaplan–Meier analysis of MC AAP patients in the present series revealed a survival probability worse than patients with IDH mutant anaplastic astrocytoma, WHO grade III and comparable to patients with IDH mutant GBM, WHO grade IV (although better than patients with IDH wildtype GBM, WHO grade IV). Notably, the comparability of the outcome data discussed above may be limited, as cases for previously performed analyses were selected by histological criteria, whereas for the present series, molecular data were additionally taken into account and may have led to a higher discrimination selectivity against other glioma entities. Another notable finding of this study was that patients with tumors, in which a characteristic MAPK pathway gene alteration was detected by the applied methods, were found to survive significantly longer than patients with tumors carrying no characteristic MAPK pathway gene alteration. The status of MAPK pathway genes in the present series was assessed by copy number profile analysis, Sanger sequencing and panel sequencing. Admittedly, these methods do not cover all rare BRAF (e.g., RNF130:BRAF, CLCN6:BRAF, MKRN1:BRAF, GNAI1:BRAF etc.) or RAF1 fusions or other rarer events. In contrast, Jones et al. detected MAPK pathway gene alterations in 96/96 PAs when applying RNA and whole-genome sequencing [25]. Therefore, it is possible that some of the MC AAP tumors without a characteristic MAPK pathway gene alteration rather harbor rare alterations that we were not able to detect using our approach. Hence, further analyses, such as RNA and whole-genome sequencing, are required to explore possible alternative alterations in the tumors of the MC AAP and, when indicated, to reappraise, whether the absence of MAPK pathway gene alterations may indeed be associated with a worse outcome.
For diagnostic practice, it is important to appreciate that the tumors allotted to the MC AAP show overlapping histological and/or molecular features to PA, pilocytic astrocytoma with anaplastic features, PXA and—to some extent—to DMG K27 and to GBM. In particular, PXAs frequently harbor alterations of BRAF (typically V600E mutation) combined with loss of CDKN2A/B [27, 57] and PAs frequently show a range of MAPK gene alterations [17, 21, 22]. Hence, distinction between the above-named entities may become challenging [31]. Figure 9 provides a suggestion of an algorithm for the diagnosis of AAP. As indicated, immunohistochemical and single parameter molecular analysis may at least help to exclude other glioma classes. Nonetheless, comprehensive molecular investigations (such as DNA methylation profiling and/or gene panel sequencing) may be necessary to definitively confirm this diagnosis.

Diagnostic considerations for the classification of anaplastic astrocytoma with piloid features (AAP): PA conventional pilocytic astrocytoma; PXA pleomorphic xanthoastrocytoma; GBM glioblastoma; DMG diffuse midline glioma; MC AAP methylation class anaplastic astrocytoma with piloid features
In conclusion, we demonstrate that MC AAP tumors are characterized by a common DNA methylation profile and recurrent alterations of NF1, BRAF or FGFR1 together with CDKN2A/B and ATRX loss as well as MGMT promoter hypermethylation. AAP most often arises as a primary tumor in the posterior fossa of young to middle-aged adults, although it may possibly also develop secondarily in the context of a lower-grade tumor. Further investigations are required, in particular on the clinical behavior of these rare tumors, as our retrospective data is rather fragmented. Nonetheless, our attempt to molecularly characterize histologically defined anaplastic pilocytic astrocytomas may provide a basis for establishing the position of the MC AAP in brain tumor classifications and may—at some stage—enable a refinement of stratification of patients for clinical trials.
References
Alpers CE, Davis RL, Wilson CB (1982) Persistence and late malignant transformation of childhood cerebellar astrocytoma. Case report. J Neurosurg 57:548–551. https://doi.org/10.3171/jns.1982.57.4.0548
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. https://doi.org/10.1093/bioinformatics/btu049
Azad A, Deb S, Cher L (2009) Primary anaplastic pilocytic astrocytoma. J Clin Neurosci 16:1704–1706. https://doi.org/10.1016/j.jocn.2009.04.012
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124:547–560. https://doi.org/10.1007/s00401-012-1016-2
Burkhard C, Di Patre PL, Schuler D, Schuler G, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H (2003) A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg 98:1170–1174. https://doi.org/10.3171/jns.2003.98.6.1170
Capper DS (2016) MolecularNeuropathology.org—the platform for next generation neuropathology. Reference set (classifier version: 11b2). https://www.molecularneuropathology.org2016. Accessed 6 Feb 2018
Capper D, Jones DTW, Sill M, Hovestadt V et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature. https://doi.org/10.1038/nature26000
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164:550–563. https://doi.org/10.1016/j.cell.2015.12.028
Collins VP, Jones DT, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 129:775–788. https://doi.org/10.1007/s00401-015-1410-7
Cyrine S, Sonia Z, Mounir T, Badderedine S, Kalthoum T, Hedi K, Moncef M (2013) Pilocytic astrocytoma: a retrospective study of 32 cases. Clin Neurol Neurosurg 115:1220–1225. https://doi.org/10.1016/j.clineuro.2012.11.009
Ebrahimi A, Skardelly M, Bonzheim I, Ott I, Muhleisen H, Eckert F, Tabatabai G, Schittenhelm J (2016) ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol Commun 4:60. https://doi.org/10.1186/s40478-016-0331-6
Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, Schackert G, Kreth FW, Pietsch T, Loffler M et al (2011) Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 129:659–670. https://doi.org/10.1002/ijc.26083
Fiechter M, Hewer E, Knecht U, Wiest R, Beck J, Raabe A, Oertel MF (2016) Adult anaplastic pilocytic astrocytoma—a diagnostic challenge? A case series and literature review. Clin Neurol Neurosurg 147:98–104. https://doi.org/10.1016/j.clineuro.2016.06.005
Gessi M, Engels AC, Lambert S, Rothamel T, von Hornstein S, Collins VP, Denkhaus D, Gnekow A, Pietsch T (2016) Molecular characterization of disseminated pilocytic astrocytomas. Neuropathol Appl Neurobiol 42:273–278. https://doi.org/10.1111/nan.12256
Groenendijk FH, Taal W, Dubbink HJ, Haarloo CR, Kouwenhoven MC, van den Bent MJ, Kros JM, Dinjens WN (2011) MGMT promoter hypermethylation is a frequent, early, and consistent event in astrocytoma progression, and not correlated with TP53 mutation. J Neurooncol 101:405–417. https://doi.org/10.1007/s11060-010-0274-x
Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS, Magrini V, Demeter R, Wylie T, Kandoth C et al (2013) Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res 23:431–439. https://doi.org/10.1101/gr.142604.112
Hasselblatt M, Riesmeier B, Lechtape B, Brentrup A, Stummer W, Albert FK, Sepehrnia A, Ebel H, Gerss J, Paulus W (2011) BRAF-KIAA1549 fusion transcripts are less frequent in pilocytic astrocytomas diagnosed in adults. Neuropathol Appl Neurobiol 37:803–806. https://doi.org/10.1111/j.1365-2990.2011.01193.x
Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, Alon N, Kahn D, Fried I, Scheinemann K et al (2011) BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17:4790–4798. https://doi.org/10.1158/1078-0432.CCR-11-0034
Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R, Cavalli FM, Ramaswamy V, Zapatka M, Reifenberger G et al (2013) Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125:913–916. https://doi.org/10.1007/s00401-013-1126-5
Hovestadt V, Zapatka M conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays. R package version 1.9.0. http://bioconductor.org/packages/conumee/. Accessed 5 May 2017
Jeuken JW, Wesseling P (2010) MAPK pathway activation through BRAF gene fusion in pilocytic astrocytomas; a novel oncogenic fusion gene with diagnostic, prognostic, and therapeutic potential. J Pathol 222:324–328. https://doi.org/10.1002/path.2780
Jones DT, Gronych J, Lichter P, Witt O, Pfister SM (2012) MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci 69:1799–1811. https://doi.org/10.1007/s00018-011-0898-9
Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932. https://doi.org/10.1038/ng.2682
Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28:2119–2123. https://doi.org/10.1038/onc.2009.73
Jones DTW, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DAK et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927-U295. https://doi.org/10.1038/ng.2682
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026. https://doi.org/10.1073/pnas.1303607110
Komine C, Watanabe T, Katayama Y, Yoshino A, Yokoyama T, Fukushima T (2003) Promoter hypermethylation of the DNA repair gene O6-methylguanine-DNA methyltransferase is an independent predictor of shortened progression free survival in patients with low-grade diffuse astrocytomas. Brain Pathol 13:176–184
Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, Dickson MA, Tap WD, Singer S, Koff A (2017) ATRX is a regulator of therapy induced senescence in human cells. Nat Commun 8:ARTN 386. https://doi.org/10.1038/s41467-017-00540-5
Krijthe JH (2015) Rtsne: T-distributed stochastic neighbor embedding using a Barnes–Hut implementation. https://github.com/jkrijthe/Rtsne. Accessed 5 May 2017
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016) World Health Organization histological classification of tumours of the central nervous system. International Agency for Research on Cancer, Lyon
Matyja E, Grajkowska W, Stepien K, Naganska E (2016) Heterogeneity of histopathological presentation of pilocytic astrocytoma—diagnostic pitfalls. A review. Folia Neuropathol 54:197–211
Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJ, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ et al (2015) DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 28:307–317. https://doi.org/10.1016/j.ccell.2015.07.012
McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N et al (2011) deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol 7:e1001138. https://doi.org/10.1371/journal.pcbi.1001138
Mellai M, Monzeglio O, Piazzi A, Caldera V, Annovazzi L, Cassoni P, Valente G, Cordera S, Mocellini C, Schiffer D (2012) MGMT promoter hypermethylation and its associations with genetic alterations in a series of 350 brain tumors. J Neurooncol 107:617–631. https://doi.org/10.1007/s11060-011-0787-y
Olar A, Wani KM, Wilson CD, Zadeh G, DeMonte F, Jones DT, Pfister SM, Sulman EP, Aldape KD (2017) Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 133:431–444. https://doi.org/10.1007/s00401-017-1678-x
Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. https://doi.org/10.1016/j.ccell.2015.04.002
Pathak P, Kumar A, Jha P, Purkait S, Faruq M, Suri A, Suri V, Sharma MC, Sarkar C (2017) Genetic alterations related to BRAF-FGFR genes and dysregulated MAPK/ERK/mTOR signaling in adult pilocytic astrocytoma. Brain Pathol 27:580–589. https://doi.org/10.1111/bpa.12444
Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, Sicotte H, Kollmeyer TM, McCoy LS, Sarkar G et al (2017) Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol 133:1001–1016. https://doi.org/10.1007/s00401-017-1690-1
Quillien V, Lavenu A, Karayan-Tapon L, Carpentier C, Labussiere M, Lesimple T, Chinot O, Wager M, Honnorat J, Saikali S et al (2012) Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, methylight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 118:4201–4211. https://doi.org/10.1002/cncr.27392
Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E et al (2011) BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res 17:3590–3599. https://doi.org/10.1158/1078-0432.CCR-10-3349
Reifenberger G, Hentschel B, Felsberg J, Schackert G, Simon M, Schnell O, Westphal M, Wick W, Pietsch T, Loeffler M et al (2012) Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer 131:1342–1350. https://doi.org/10.1002/ijc.27385
Reuss DE, Kratz A, Sahm F, Capper D, Schrimpf D, Koelsche C, Hovestadt V, Bewerunge-Hudler M, Jones DT, Schittenhelm J et al (2015) Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol 130:407–417. https://doi.org/10.1007/s00401-015-1454-8
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V et al (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. https://doi.org/10.1007/s00401-014-1370-3
Rodriguez EF, Scheithauer BW, Giannini C, Rynearson A, Cen L, Hoesley B, Gilmer-Flynn H, Sarkaria JN, Jenkins S, Long J et al (2011) PI3K/AKT pathway alterations are associated with clinically aggressive and histologically anaplastic subsets of pilocytic astrocytoma. Acta Neuropathol 121:407–420. https://doi.org/10.1007/s00401-010-0784-9
Rodriguez FJ, Scheithauer BW, Burger PC, Jenkins S, Giannini C (2010) Anaplasia in pilocytic astrocytoma predicts aggressive behavior. Am J Surg Pathol 34:147–160. https://doi.org/10.1097/PAS.0b013e3181c75238
Rohrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz A, Reuss J, Hovestadt V, Jones DT, Bewerunge-Hudler M et al (2016) Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol 131:877–887. https://doi.org/10.1007/s00401-016-1540-6
Roth JJ, Santi M, Rorke-Adams LB, Harding BN, Busse TM, Tooke LS, Biegel JA (2014) Diagnostic application of high resolution single nucleotide polymorphism array analysis for children with brain tumors. Cancer Genet 207:111–123. https://doi.org/10.1016/j.cancergen.2014.03.002
Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D, Capper D, Koelsche C, Korshunov A, Wiestler B et al (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. https://doi.org/10.1007/s00401-015-1519-8
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, Okonechnikov K, Koelsche C, Reuss DE, Capper D et al (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18:682–694. https://doi.org/10.1016/S1470-2045(17)30155-9
Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S et al (2012) Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231–1235. https://doi.org/10.1126/science.1220834
Stuer C, Vilz B, Majores M, Becker A, Schramm J, Simon M (2007) Frequent recurrence and progression in pilocytic astrocytoma in adults. Cancer 110:2799–2808. https://doi.org/10.1002/cncr.23148
Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF et al (2014) Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer 14:92–107. https://doi.org/10.1038/nrc3655
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I et al (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072. https://doi.org/10.1016/j.cell.2016.01.015
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Theeler BJ, Yung WK, Fuller GN, De Groot JF (2012) Moving toward molecular classification of diffuse gliomas in adults. Neurology 79:1917–1926. https://doi.org/10.1212/WNL.0b013e318271f7cb
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164. https://doi.org/10.1093/nar/gkq603
Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B, Becker AJ, Weber-Mangal S, Jauch A, Radlwimmer B et al (2007) Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene 26:1088–1097. https://doi.org/10.1038/sj.onc.1209851
Yong EX, McKelvie P, Murphy M, Wang YY (2014) Anaplastic pilocytic astrocytoma. J Clin Neurosci 21:1993–1996. https://doi.org/10.1016/j.jocn.2014.02.014
Zacher A, Kaulich K, Stepanow S, Wolter M, Kohrer K, Felsberg J, Malzkorn B, Reifenberger G (2017) Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol 27:146–159. https://doi.org/10.1111/bpa.12367
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I et al (2013) Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45:602–612. https://doi.org/10.1038/ng.2611
Acknowledgements
We thank the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ) for the performance of methylation analyses and Dr. Stefan Uhrig and Dr. Gnana Prakash Balasubramanian for the help with the detection of gene fusions in the gene panel sequencing raw data. Part of this work was undertaken at UCLH/UCL which received funding from the National Institute of Health Research (NIHR) (Sebastian Brandner). We thank the DKFZ Heidelberg Center for Personalized Oncology (DKFZ-HIPO) for technical support and funding through HIPO_036. In other parts, this work was supported by an Illumina Medical Research Grant and the German Cancer Consortium (DKTK) joint funding project ‘Next Generation Molecular Diagnostics of Malignant Gliomas’.
Author information
Authors and Affiliations
Corresponding author
Additional information
David T. W. Jones, Andreas von Deimling and David Capper share senior authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Reinhardt, A., Stichel, D., Schrimpf, D. et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136, 273–291 (2018). https://doi.org/10.1007/s00401-018-1837-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1837-8