
Abstract
It has been reported that the development of TDP-43 pathology in cases of Lewy body disease (LBD) might be associated with the severity of tau pathology. However, the impact of α-synuclein pathology on TDP-43 accumulation in LBD remains unclear. To clarify whether α-synuclein pathology has an effect on TDP-43 accumulation, independent of tau pathology, we examined by immunohistochemistry 56 cases of LBD using a phosphorylation-dependent TDP-43 antibody. The frequency of TDP-43 pathology in all LBD cases was 18% (10/56). In 37 LBD cases with no or low tau burden (LBD-Ltau; Braak NFT stages 0-II), the frequency of TDP-43 pathology was 19% (7/37). The frequency of TDP-43 pathology in diffuse neocortical type LBD-Ltau cases was 36% (4/11), which was higher than those in limbic and brain stem-predominant types (11–14%). The amygdala and entorhinal cortex were the most frequently affected sites of TDP-43 pathology in LBD-Ltau cases. In LBD-Ltau cases, the proportion of diffuse neocortical type LBD was higher in the TDP-43-positive cases, than that in TDP-43-negative cases (57 vs. 23%). In all LBD cases, α-synuclein pathology in the temporal cortex was significantly more severe in TDP-43-positive cases, and significantly correlated with the severity of TDP-43 pathology in the amygdala. In a multivariate model, the presence of severe α-synuclein pathology was significantly associated with the development of TDP-43 pathology independent of age at death and tau pathology. In the amygdala, TDP-43 was often colocalized with α-synuclein or tau. Given these findings, we suggest that α-synuclein pathology is associated with TDP-43 accumulation in LBD cases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The transactivation-responsive DNA-binding protein of Mr 43 kDa, TDP-43, is a major component of ubiquitin-positive and tau-negative inclusions in the frontotemporal cortex and motor neurons in frontotemporal lobar degeneration (FTLD-U) and in amyotrophic lateral sclerosis (ALS), and is considered to play an essential pathogenic role in these diseases, now called TDP-43 proteinopathies [3, 6, 7, 29]. However, abnormal TDP-43 accumulations have been demonstrated in cases of Alzheimer’s disease (AD) [1, 2, 15], ALS/parkinson–dementia complex of Guam (ALS/PDC of Guam) [9, 12], argyrophilic grain disease (AGD) [8], corticobasal degeneration (CBD) [32], and progressive supranuclear palsy (PSP) [34]. Additionally, some (but not all) studies have supported the possibility that the severity of tau pathology is associated with TDP-43 accumulation in AD [1, 2], AGD [8], and PSP [34].
A few studies have demonstrated a concurrent TDP-43 pathology in some cases with Lewy body disease (LBD), including ones with Parkinson’s disease (PD), Parkinson’s disease with dementia (PDD), and dementia with Lewy bodies (DLB). The reported frequencies of TDP-43 pathological changes in several LBD series ranged from 19 to 60% [2, 14, 28]. However, most LBD cases have variable degrees of AD-type pathology [11, 17, 21–23, 27, 30, 33]. Indeed, in the earliest (and largest) study that examined TDP-43 pathology in LBD, approximately 50% of 180 LBD cases had moderate to severe tau pathology, and a higher frequency of TDP-43 pathology was observed in cases with a more severe Braak NFT stage score [28]. In a recent study, about 70% of TDP-43-positive LBD cases had moderate to severe tau pathology (Braak NFT stages III–VI) [2]. Nonetheless, somewhat unexpectedly, it has never been examined whether the development of TDP-43 pathology in LBD is influenced by α-synuclein pathology, or can simply be explained by the effect of concurrent tau pathology. Higashi et al. [14] reported no significant difference in the severity of α-synuclein pathology between DLB cases with or without TDP-43 pathology, and AD cases with or without TDP-43 pathology. However, the number of subjects in the study was small (11 DLB cases including 5 TDP-43-positive cases, and 15 AD cases including 5 TDP-43-positive cases), and the influence of tau pathology was not compensated for.
The principal aim of this study was to investigate whether the presence of α-synuclein pathology is associated with TDP-43 accumulation in LBD. To address this, we revisited the frequency and distribution of TDP-43 pathology using a phosphorylation-dependent TDP-43 antibody in LBD cases with no or low tau burden (corresponding to Braak NFT stages 0–II [4]) (i.e., LBD-Ltau cases) and LBD cases with more severe tau burden (Braak NFT stages III–VI) (LBD-Htau cases). Secondly, we compared the severities of α-synuclein and tau pathologies between LBD cases with and without TDP-43 accumulation, and also examined the correlation between the severity of TDP-43 pathology and that of α-synuclein or tau pathology. Thirdly, we examined the frequencies of TDP-43 pathology in three subtypes of LBD (i.e., brain stem-predominant type, limbic type, and diffuse neocortical type [27]) concentrating especially on the LBD-Ltau cases, and asking whether the severity of α-synuclein pathology was independently associated with the development of TDP-43 pathology in all LBD cases using multivariate models. Finally, we performed double immunofluorescence labeling and biochemical examination in order to further understand the pathogenic mechanism underlying TDP-43 accumulation in LBD.
Materials and methods
Subjects
All of the available pathologically confirmed LBD cases (n = 56) in the UK Parkinson’s Disease Society Tissue Bank, as well as pathologically normal controls (n = 4), were examined in this study. The clinical diagnosis in these cases was LBD (i.e., 29 cases of PD, 51.8%; 23 cases of PDD, 41.1%; and 4 cases of DLB, 7.1%). The clinical diagnosis of PDD was based on motor impairment preceding cognitive impairment by at least 1 year [27]. The most frequent pathological subtype of LBD in our series was limbic type (51.8%), followed by the diffuse neocortical type (30.4%) and lastly the brainstem-predominant type (17.9%). No cases having other degenerative diseases, such as PSP, CBD, and multiple system atrophy, were included in this study. The proportion of LBD cases with severe tau pathology in this series was low: 37 cases (66% of all LBD cases) had no or low tau burden corresponding to Braak NFT stages 0–II (i.e., were LBD-Ltau cases): 30 cases (53.6%) corresponded to Braak NFT stage II, six cases (10.7%) were Braak NFT stage I, and only one case completely lacked tau pathology (1.8%). The other 19 cases (34%) had a higher tau burden corresponding to Braak NFT stages III–VI (i.e., LBD-Htau cases): ten cases (17.9%) corresponded to Braak NFT stage III, four cases (7.1%) had stage IV, three cases (5.4%) had stage V, and two cases (3.6%) had stage VI. Thirty-eight cases (68% of all LBD cases) had various degrees of Aβ deposits in the hippocampus and/or temporal cortex. Argyrophilic grains were found in two LBD cases. All brains had been collected with Local Research Ethical Committee approval. Relevant clinical and pathological features for all 56 LBD cases are shown in Table 1.
Immunohistochemistry
Paraffin sections were cut at 6 μm thickness, to include the amygdala, entorhinal cortex, hippocampus, and occipitotemporal cortex, from all LBD cases and immunostained with antibodies against phosphorylated TDP-43 (pAb pS409/410, rabbit, polyclonal, 1:1,000 [13]), phosphorylated tau (AT8, mouse, monoclonal, 1:3,000, Innogenetics, Ghent, Belgium), phosphorylated α-synuclein (#1175, rabbit, polyclonal, 1:1,000, [30]), and Aβ (4G8, mouse, monoclonal, 1:2,000, Covance Research Products Inc., Dedham, MA). Deparaffinized sections were incubated with 1% H2O2 in methanol for 20 min to eliminate endogenous peroxidase activity. When using anti-α-synuclein and anti-TDP-43 antibodies, sections were pretreated in a microwave oven for 5 min in 10 mM sodium citrate buffer, pH 6.0, at 100°C to enhance immunoreaction. For Aβ immunostaining, sections were incubated in 95% formic acid for 5 min. No pretreatment was performed for AT8 immunostaining. After blocking with 10% normal serum, sections were incubated for 1 h at room temperature with one of the primary antibodies. After three 5-min washes in PBS, sections were incubated in biotinylated secondary antibody for 30 min, and then in avidin–biotinylated horseradish peroxidase complex (ABC Elite kit, Vector, Burlingame, CA, USA) for 30 min. The peroxidase labeling was visualized with 0.2% 3,3′-diaminobenzidine (DAB) as chromogen. Sections were lightly counterstained with hematoxylin.
Semiquantitative assessment
TDP-43, α-synuclein, tau, and Aβ pathologies in the amygdala, anterior and posterior portions of the entorhinal cortex, hippocampal dentate gyrus, CA1, 2, 3, and 4 regions, subiculum, fusiform gyrus, and occipitotemporal gyrus were semiquantitatively evaluated using the following grading system blinded to any clinical or pathological information:
-
(a)
The total number of TDP-43-positive neuronal cytoplasmic inclusions (NCIs) in each anatomical region was assessed as follows: −, no lesion; +, one inclusion; ++, two or three inclusions; +++, four or five inclusions; ++++, 6–10 inclusions; +++++, 11 or over inclusions. The topographic distribution of TDP-43 pathological changes was assessed using the following system, which was similar to that reported by Amador-Ortiz et al. [1]—the amygdala type: inclusions were present only in the amygdala; the limbic type: inclusions extend to the amygdala, hippocampal dentate gyrus, CA1-4, entorhinal cortex, and fusiform gyrus, but not in the occipitotemporal gyrus; the temporal type: inclusions are present in the limbic system and also in the occipitotemporal gyrus.
-
(b)
The LBD cases were classified, irrespective of the presence or absence of dementia, into brain stem-predominant type, limbic type, and diffuse neocortical type, according to the distribution of α-synuclein pathology as recommended by the Third Consensus Guideline for DLB [27]. In addition, the severity of α-synuclein pathology in the substantia nigra, amygdala, and temporal cortex was semiquantitatively assessed at ×100 magnification using the following method, again fundamentally consistent with protocols of the Third Consensus Guideline for DLB [27]: grade 1, one Lewy body (LB) or Lewy neurites (LNs) per few fields; grade 2, one to three LBs and sparse LNs per one field; grade 3, four to ten LBs and scattered LNs per one field; grade 4, over 11 LBs and LNs per one field.
-
(c)
Tau-positive neuronal inclusions were counted at ×100 magnification: 0, no tau-positive lesions; 1, one neuronal inclusion per few microscopic fields; 2, one to three inclusions in every field; 3, 4–30 inclusions in every field; 4, over 30 inclusions associated with numerous neurites in every field. The distribution of tau pathology in LBD cases was assessed according to Braak NFT stage on AT8 immunostained sections [4].
-
(d)
Aβ deposits were counted at ×100 magnification: 0, no Aβ deposits; 1, two to three Aβ plaques in each field; 2, 4–10 Aβ plaques in each field; 3, 11–20 Aβ plaques in each field; 4, more than 20 Aβ deposits in each field.
Hippocampal sclerosis (HS) was defined by neuronal loss with gliosis in the hippocampal CA1 and/or subiculum, with relatively preserved neurons in CA2, 3, and 4 regions and absence of intracellular and extracellular NFTs, or ischemic changes that might explain neuronal loss in the CA1 and subiculum. HS was assessed on hematoxylin–eosin stained sections blind to any clinical or pathological information.
Statistical analysis
The Mann–Whitney U test and Fisher’s exact test were used to compare the demographic and pathological data between two groups. Correlations between (a) the rating of TDP-43 pathology in the amygdala and clinical variables, (b) the rating of TDP-43 pathology and that of α-synuclein, tau, or Aβ pathology in each anatomical region, and (c) the rating of TDP-43 pathology in the amygdala and that of α-synuclein or tau pathology in each region were assessed by Spearman’s rank-order correlation test. Multiple logistic regression models were used to assess the influence of predictor variables (age at death, the severities of tau and α-synuclein pathologies) on the occurrence of TDP-43 pathology. The effects were described as odds ratios and 95% confidence interval (CI). Statistical analysis was performed using Excel, Stat View version J-4.5, and SPSS 10.0J. A P value <0.05 was accepted as significant; however, in analyses of comparisons between two groups and correlations between two variables, a P value <0.01 was accepted as significant to interpret the results with caution because multiple tests have been done.
Confocal laser scanning microscopy
Double-labeling immunofluorescence was performed with the combination of (a) phosphorylation-dependent rabbit polyclonal anti-TDP-43 (pAb pS409/410, 1:1,200 [13]) and anti-tau antibodies (AT8, mouse, monoclonal, 1:500, Innogenetics, Ghent, Belgium), and (b) phosphorylation-dependent mouse monoclonal anti-TDP-43 (mAb pS409/410, 1:1,200 [16]) and phosphorylation-dependent anti-α-synuclein antibodies (#1175, rabbit, polyclonal, 1:1,000, [30]). Sections from the amygdala in LBD cases with TDP-43 pathology were pretreated by heating in a microwave oven for 5 min in 10 mM sodium citrate buffer, pH 6.0, at 100°C, allowed to cool then permeabilized with 0.2% (v/v) Triton X-100 in phosphate buffered saline (PBS). Following washing in PBS, non-specific antibody binding was blocked with normal sera and sections were incubated with a mixture of the two primary antibodies for 1 h at room temperature. After washing in PBS, sections were incubated with fluorescence-labeled secondary antibodies [AlexaFluor 488 anti-rabbit IgG (1:200) and AlexaFluor 555 anti-mouse IgG (1:200), Molecular Probes, Invitrogen, Paisley, UK]. After washing with PBS, sections were incubated with Toto-3 Iodide (Molecular Probes, Invitrogen, Paisley, UK) with 1 mg/ml RNase (Roche Diagnostics GmbH, Manheim, Germany) at 37°C. To quench (lipofuscin) autofluorescence, sections were incubated in 0.1% Sudan Black B for 10 min at room temperature and washed with 0.1% Triton X-PBS for 30 min. Sections were coverslipped with Vectashield mounting media (Vector Laboratories Inc., Burlingame, CA). Images were collected on a Leica TCS SP5 AOBS upright confocal (Leica Microsystems, Milton Keynes, UK) using the 488 nm (19%), 543 nm (30%) and 633 nm (60%) laser lines. To eliminate cross-talk between channels, the images were collected sequentially.
Immunoblotting
Frozen tissues from the amygdala, hippocampus, and frontal and temporal cortex from two LBD cases (one TDP-43-positive and one TDP-43-negative case), one FTLD-TDP case as a positive control, and one pathologically normal control case were prepared for western blotting according to methods previously described by Neumann et al. [29]. Briefly, fresh frozen brain was homogenized in low salt (LS) buffer containing 10 mM Tris pH 7.5, 5 mM EDTA pH 8.0, 1 mM DTT, 10% (w/v) sucrose and Roche complete EDTA free protease inhibitor. Homogenates were sequentially extracted with increasing strength buffers [Triton X-100 buffer (LS buffer + 1% Triton X-100 + 0.5 M NaCl), Triton X-100 buffer with 30% sucrose to float myelin, Sarkosyl buffer (LS buffer + 1% N-lauroyl-sarcosine + 0.5 M NaCl)]. Detergent insoluble pellets were extracted in 0.25 ml/g urea buffer (7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 30 mM Tris–HCl pH 8.5, Roche complete EDTA free protease inhibitor). Prior to SDS-PAGE immunoblot analysis, urea fractions were added in 1:1 ratio to SDS sample buffer (10 mM Tris pH 6.8, 1 mM EDTA, pH 8.0, 40 mM DTT, 1% SDS, 10% sucrose, 0.01% bromophenol blue). Protein was resolved on 12% Tris–glycine SDS-PAGE gels along with size standard (Bio-Rad kaleidoscope broad-range marker; BioRad, Hercules, CA). Proteins were transferred onto nitrocellulose membrane (Hybond ECL, GE Life Sciences, UK) and blocked overnight at 4°C in 5% (w/v) milk solution [5% powdered milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T)]. Membranes were incubated in phosphorylation-dependent mouse monoclonal antibody (mAb pS409/410, mouse, 1:1,000 [16]) for 1 h at room temperature followed by HRP-conjugated goat anti-mouse secondary antibody (Santa Cruz Biotechnology Inc, CA). Antibodies were visualized by incubating in enhanced chemiluminescent reagent (ECL, GE Life Sciences) and imaged using the ImageQuant 350 system fitted with a F0,95 25 mm Fixed Lens (GE Healthcare, Life Sciences, UK). TDP-43-probed membranes were exposed for 5 min at different timeframes to obtain multiple images of differing intensity. Images were processed using ImageQuant TL software (GE Healthcare, Life Sciences, UK).
Results
Frequency and distribution of TDP-43 pathology in all LBD cases
Of the 56 LBD cases, 10 (17.9%) had TDP-43-positive neuronal intracytoplasmic inclusions (NCIs) (Table 1). The amygdala (all 10 TDP-43 positive cases) was most frequently affected by TDP-43 pathology, followed by the anterior portion of the entorhinal cortex (7/10 cases), hippocampal dentate gyrus (3 cases), subiculum (3 cases), and CA1, fusiform gyrus, and occipitotemporal gyrus (2 cases for each) (Table 2; Fig. 1). The distribution of TDP-43 pathology was the amygdala type in one case, the limbic type in seven cases, and the temporal type in two cases. No neuronal intranuclear inclusions were noted in any LBD case.

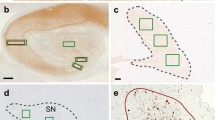
Phosphorylated TDP-43 pathology in LBD-Ltau cases. TDP-43-positive NCIs in the entorhinal cortex (a), amygdala (b, c), and CA1 (d). TDP-43-positive dystrophic neurites are also scattered in the amygdala (e). In some cases, abundant fine and short threads-like structures are also seen in the CA1 to subiculum (f, g). pAb pS409/410 immunohistochemistry. a, c, d, g Diffuse neocortical type LBD-Ltau cases (Braak NFT stage II), b, e, f limbic type LBD-Ltau cases (Braak NFT stage I). Scale bars a 100 μm, b–g 20 μm
α-Synuclein and tau pathologies in LBD cases with and without TDP-43 pathology
Clinical and pathological features in those LBD cases with and without TDP-43 pathology are shown in Table 1. There was no statistically significant difference in the sex ratio, mean age at onset, age at death, disease duration, or frequency of dementia between these two groups. There was no significant correlation between demographic variables and the rating of TDP-43 pathology in the amygdala. None of our LBD cases, including TDP-43-positive cases, had significant neuronal loss in the hippocampal CA1 or subiculum consistent with the definition of HS.
α-Synuclein pathology in the 10 TDP-43-positive cases was more widely distributed than that in TDP-43-negative cases. The diffuse neocortical type of LBD was the most common pathological subtype in the TDP-43-positive cases (six cases), followed by limbic type and brainstem-predominant type (two cases each) (Fig. 2a). In contrast, in the TDP-43-negative LBD cases, the limbic type was most frequent (58.7%), while only 23.9% cases had diffuse neocortical type (Fig. 2a). The frequency of diffuse neocortical type cases in the TDP-43-positive cases tended to be higher than that in the TDP-43-negative cases (Mann–Whitney U test, P = 0.052). Consistent with these results, was the observation that the rating of α-synuclein pathology in the temporal cortex in the TDP-43-positive cases was significantly more severe than that in the TDP-43-negative cases (Mann–Whitney U test, P = 0.003; Fig. 2b). There was no significant difference in the rating of α-synuclein pathology, in either the substantia nigra or the amygdala, between the TDP-43-positive and TDP-43-negative cases. The Spearman rank correlation coefficient showed a moderate correlation between the ratings for α-synuclein pathology in the temporal cortex and TDP-43 pathology in the amygdala (Spearman rho = 0.398, P < 0.01). In any other regions, there was no significant correlation between the ratings for TDP-43 and α-synuclein pathologies, and Spearman rho ranged from −0.087 to 0.122.

α-Synuclein pathology in all LBD cases with and without TDP-43 pathology. a The distribution of pathological subtypes of LBD in TDP-43-positive and TDP-43-negative groups. The frequency of diffuse neocortical type in TDP-43-positive LBD cases tends to be higher than that in TDP-43-negative LBD cases (P = 0.052). The number of cases in each group is shown in brackets. b The rating of α-synuclein pathology in TDP-43-positive and TDP-43-negative LBD cases. α-Synuclein pathology in the temporal cortex in TDP-43-positive cases was significantly more severe than that in TDP-43-negative cases (P = 0.003)
The ratings for tau pathology in the hippocampal dentate gyrus in the TDP-43-positive cases tended to be higher than those in the TDP-43-negative cases (Mann–Whitney U test, P = 0.037). Likewise, although not significantly, Braak NFT stage in the TDP-43-positive cases also tended to be higher than that in the TDP-43-negative cases (Fig. 3). Although not significant, a moderate correlation was observed between the ratings for tau pathology in the hippocampal dentate gyrus and those for TDP-43 pathology in the amygdala (Spearman rho = 0.301, P < 0.05). In any other regions, there was no significant correlation between the ratings for TDP-43 and tau pathologies, and Spearman rho ranged from −0.092 to 0.178.

Braak NFT stage in LBD cases with and without TDP-43 pathology. Tau pathology in TDP-43-positive LBD cases tended to be more severe than that in TDP-43-negative LBD cases. The number of cases in each group is shown in brackets
Ratings for Aβ pathology were not significantly different between the TDP-43-positive and TDP-43-negative LBD cases. Spearman correlation coefficients did not indicate any significant correlation between the severities of Aβ and TDP-43 pathologies in any region, and Spearman rho ranged from 0.103 to 0.227.
Relationship between α-synuclein and TDP-43 pathologies in LBD-Ltau and LBD-Htau cases
The relationship between α-synuclein and TDP-43 pathologies in LBD-Ltau (Braak NFT stages 0–II) and LBD-Htau cases (Braak NFT stages III–VI) was examined separately. The sex ratio, mean age at onset, age at death, disease duration, frequency of dementia were not significantly different between the TDP-43-positive and TDP-43-negative cases in the LBD-Ltau cases, as well as in the LBD-Htau cases (Table 3).
In the LBD-Ltau cases with TDP-43 pathology, the most frequent LBD subtype was the diffuse neocortical type (57.1%), followed by limbic (28.6%) and brain stem-predominant (14.3%) types (Fig. 4). In contrast, in LBD-Ltau cases without TDP-43 pathology, the limbic type was most frequent (56.7%), while the diffuse neocortical type was seen in only 23.3% cases and brain stem-predominant type in 20.0%. In the LBD-Ltau group, the rating of α-synuclein pathology in the temporal cortex in the TDP-43-positive cases tended to be higher than that in the TDP-43-negative cases (Mann–Whitney U test, P = 0.042). Although case numbers were small, a similar trend was seen in the LBD-Htau cases: 2 of 3 TDP-43-positive LBD cases were diffuse neocortical type, while only 4 of 16 TDP-43-negative cases were this subtype (67 vs. 25%).

Distribution of LBD subtypes in LBD-Ltau cases (Braak NFT stages 0–II). Diffuse neocortical type in TDP-43-positive cases was more frequent than that in TDP-43-negative cases. The number of cases in each group is shown in brackets
Both in LBD-Ltau or in LBD-Htau cases, the ratings for tau and Aβ pathologies were not significantly different between TDP-43-positive and TDP-43-negative cases, in any region.
Frequency of TDP-43 pathology by clinical and pathological subtypes of LBD
There was no clear relationship between the occurrence of TDP-43 pathology and clinical phenotypes of LBD in our series: TDP-43 pathology was found in 7 of 29 PD cases (24.1%), 2 of 23 PDD cases (8.7%), and 1 of 4 DLB cases (25%). The overall frequency of TDP-43 pathology was 11.1% in LBD cases with dementia and 24.1% in LBD cases without it. However, in LBD-Ltau cases (Braak NFT stages 0–II), the duration from disease onset to the development of dementia in TDP-43-positive cases tended to be shorter than that in TDP-43-negative cases (2.0 ± 1.7 vs. 11.9 ± 7.0 years, P = 0.046, Mann–Whitney U test).
In contrast to clinical phenotypes, there was a trend for TDP-43 pathology to be more frequently present in cases with severe α-synuclein pathology. In LBD-Ltau cases, TDP-43 pathology was noted in 4 of 11 diffuse neocortical type LBD-Ltau cases (36.4%), whereas it was only present in 2 of 19 cases of limbic type LBD (10.5%) and in 1 of 17 brain stem-predominant type LBD (14.3%) cases. In LBD-Htau cases, 2 of the 5 diffuse neocortical type cases with severe tau pathology (Braak NFT stages V–VI) also had TDP-43 pathology. The overall frequency of TDP-43 pathology in diffuse neocortical type LBD cases was 35.3% (6 of 17 cases).
Effects of α-synuclein and tau pathologies on development of TDP-43 pathology
A multiple logistic regression model was used to evaluate whether pathological subtypes of LBD, Braak NFT stage, and age at death could be used as possible predictors for the development of TDP-43 pathology. After combining categories in which the number of cases was small (Braak NFT stages 0–II or Braak NFT stages III–VI) and pathological subtype of LBD (diffuse neocortical type or others) data were submitted as binary variables into the model. The presence of diffuse neocortical type of LBD was the only significant independent predictor of the development of TDP-43 pathology (odds ratio 7.6, 95% CI 1.46–39.1, P = 0.016).
Multiple logistic regression analysis was also used to examine whether the ratings of the severities of α-synuclein and tau pathologies in the amygdala and age at death were predictors of the development of TDP-43 pathology. Again, after combining categories in which the number of cases was small, the ratings of α-synuclein (grades 1–3 or grade 4) and tau pathologies (Braak NFT stages 0–II or Braak NFT stages III–IV) were submitted as binary variables into the model. However, neither of these variables predicted the development of TDP-43 pathology, although the odds ratio of severe α-synuclein pathology in the amygdala was high (odds ratio 3.5, 95% CI 0.71–17.1, P = 0.122).
Double-labeling confocal microscopy of phosphorylated TDP-43, tau, and α-synuclein
In the amygdala, TDP-43 accumulation was often colocalized with α-synuclein accumulation in NCIs and dystrophic neurites (Fig. 5a–i). TDP-43 was also often colocalized with tau labeling (Fig. 5j–o), but there were also some TDP-43-positive α-synuclein-negative lesions (Fig. 5d–i) and TDP-43-positive tau-negative lesions (Fig. 5j–l). In the hippocampal granular cells, TDP-43 and tau were only rarely colocalized (data not shown).

Confocal double immunofluorescence of the combination of α-synuclein (a, d, g) and TDP-43 (b, e, h), and the combination of TDP-43 (j, m) and tau (k, n) in the amygdala in LBD cases. Blue fluorescence in merged images (c, f, i, l, o) are nuclei. a–c TDP-43 (red) is colocalized with α-synuclein (green) in an inclusion. d–f In a left inclusion, α-synuclein (green) is colocalized with TDP-43 (red). However, the right TDP-43-positive inclusion (arrow) shows only faint α-synuclein immunolabeling (green). g–i A left horseshoe-shaped inclusion is stained only with an α-synuclein antibody, while a right neuron shows diffuse cytoplasmic TDP-43 labeling (red) without α-synuclein immunoreactivity. j–o TDP-43 (green) is often colocalized with tau labeling (red) in cytoplasmic inclusions. There are a few TDP-43-positive tau-negative lesions (l, green). a–i #1175 and mAb pS409/410 double immunofluorescence in a LBD-Ltau case (Braak NFT stage II), j–o pAb pS409/410 and AT8 double immunofluorescence in a LBD-Htau case (Braak NFT stage VI). Scale bars a–c 7.5 μm, d–f 7.5 μm, g–i 7.5 μm, j–l 10 μm, m–o 25 μm
Biochemical analysis of TDP-43 in LBD cases
Immunoblot analysis of the sarkosyl-insoluble urea-soluble fraction using mAb pS409/410 in LBD cases with TDP-43 pathology demonstrated distinct bands at approximately 45 and 25 kDa, as well as high molecular weight (HMW) smears (Fig. 6, lanes 3 and 4) similar to those seen in the FTLD-TDP case (lane 6). These pathological bands and the HMW smear were not seen in the LBD cases without TDP-43 pathology (lanes 1 and 2) or in the normal control cases (lane 5).

Immunoblot analysis of the sarkosyl-insoluble fraction in representative LBD cases with phosphorylation-dependent monoclonal anti-TDP-43 antibody (mAb pS409/410). The approximately 45 and 25 kDa fragments, as well as smears are strongly labeled in a LBD case with TDP-43 pathology (lanes 3 and 4) and a FTLD-TDP case (lane 6). These 45 and 25 kDa bands and smears were not labeled in any other cases without detectable TDP-43 pathology by immunohistochemistry (lanes 1, 2, and 5). Normal 43 kDa TDP-43 is not stained by this phosphorylation-dependent antibody in any cases. LBD Lewy body disease, NC normal control, AM amygdala, HP hippocampus, F frontal cortex, T temporal cortex, IHC pAb pS409/410 immunohistochemistry
Discussion
This is the first study demonstrating a high frequency of TDP-43 pathology in LBD-Ltau cases (Braak NFT stages 0–II). The overall frequency of TDP-43 pathology was 19%, and the frequency of TDP-43 pathology in diffuse neocortical type LBD cases was as much as 36%, which was higher than those in other LBD subtypes (11–14%). In all LBD cases in this study, even in LBD-Ltau cases, the proportion of diffuse neocortical type LBD cases among the TDP-43-positive cases was approximately 1.5 times higher than that in the TDP-43-negative cases, and multivariate analysis demonstrated that severe α-synuclein pathology was a predictor of TDP-43 accumulation in LBD independent of age at death and tau pathology. Double immunofluorescence demonstrated that TDP-43 was often colocalized with α-synuclein or tau in the amygdala. These findings suggest that α-synuclein may play some role in the process associated with the development of TDP-43 pathology in LBD cases.
Previous data regarding TDP-43 accumulation in LBD cases are limited. In an early study by Nakashima-Yasuda et al. [28], the overall frequency of TDP-43 pathology in LBD cases was reported to be 18.9%. This frequency appears to be similar to that in all LBD cases examined in our study (17.9%), even though these authors employed a ‘conventional’ phosphorylation-independent TDP-43 antibody. However, these frequencies cannot be directly compared, because the pathological backgrounds between two LBD series may be different. For example, the degree of tau pathology in the Nakashima-Yasuda series tended to be more severe than that in the present series: the proportion of LBD cases having severe tau pathology (Braak NFT stages V–VI) being 21% (38 of 180 LBD cases), far higher than that in our LBD series (8.9%). Conversely, the proportion of cases of Braak NFT stages 0–II in the Nakashima-Yasuda series was about 50% (91 of 180 LBD cases), this being smaller than that in our series (66.1%). Considering that several studies have suggested a possible relationship between TDP-43 accumulation and the severity of tau pathology in several tauopathies [2, 8, 28], it is plausible that the differences regarding the degree of tau pathology might have influenced the overall frequency of TDP-43 pathology in LBD series. More recently, Arai et al. [2], using the same phosphorylation-dependent TDP-43 antibody employed in the present study, reported TDP-43 pathology in up to 56% of DLB and DLB + AD cases. Although Arai et al. did not present detailed data regarding tau and α-synuclein pathologies, the degree of tau pathology, at least, in their TDP-43-positive LBD cases tended to be more severe than that in our TDP-43-positive cases: 43% of their TDP-43-positive LBD cases was classified as having severe tau pathology of Braak NFT stages V–VI (compared to 20% in the present TDP-43-positive cases). Because of the relative paucity of cases having severe tau pathology of Braak NFT stages V–VI in our series (2 of 5 cases, 40%), it is difficult to discuss about the significance of the frequency of TDP-43 pathology in this subpopulation of LBD cases. However, similar frequencies have been observed in some subgroups in Nakashima-Yasuda et al. [28] series where the pathological background may be similar to that in our series. These authors reported that TDP-43 pathology in 47% of LBD cases of Braak NFT stages V–VI (the severity of α-synuclein pathology in this group was not shown), and in 31% of DLB + AD cases (a high and intermediate likelihood for both DLB and AD pathology [27, 31]). We could not fully examine the differences of the severity and distribution of TDP-43 pathology between diffuse neocortical type of LBD cases with and without severe tau pathology, because the number of cases having severe tau pathology was small. The data regarding the difference of TDP-43 pathology between these two groups, including the presence or absence of TDP-43 accumulation in the frontal and occipital cortices, may provide clues to understand the impacts of not only α-synuclein but also tau accumulations on TDP-43 accumulation in LBD cases.
Whereas the degree of α-synuclein pathology is highly variable among LBD cases, there was few previous data available regarding the relationship between the severity of α-synuclein pathology and the development of TDP-43 pathology. In the light of present results, the severity of α-synuclein pathology may be a potential factor for the development of TDP-43 pathology. We suggest that the severity of α-synuclein pathology should be considered when interpreting the frequency of TDP-43 pathology in LBD cases, and probably, in other pathological conditions as well. It is notable that TDP-43 pathology was frequently found in our LBD cases even when severe tau pathology did not coexist, especially in cases of the diffuse neocortical type. Although inconsistent with findings of an early study in which none of ten LBD-Ltau cases had TDP-43 pathology [28], Higashi et al. [14] reported TDP-43 pathology in 3/7 LBD-Ltau cases using a phosphorylation-independent antibody and Arai et al. [2] reported that all of four LBD-Ltau cases in their series had variable degrees of TDP-43 pathology. Our present results agree with these findings and suggest a possible association of α-synuclein and TDP-43 accumulations in LBD-Ltau cases.
On the other hand, in a study by Josephs et al. [20] in which 84 AD cases were examined, multivariate analysis did not demonstrate any significant effect of the presence of α-synuclein pathology on the development of TDP-43 accumulation in AD. In their AD series, although the prevalence of α-synuclein pathology was only 25%, the frequency of α-synuclein pathology in TDP-43-positive cases was significantly higher than in TDP-43-negative cases (38 vs. 18%). No detailed data about the degree of α-synuclein pathology in this series was presented. In the context of present findings, if the degree of α-synuclein pathology in this series [20] was mild, the effect of α-synuclein pathology on the development of TDP-43 accumulation would not likely be demonstrated.
Although multivariate analysis in our study failed to demonstrate a significant association between tau pathology and TDP-43 accumulation, the result does not necessarily deny the possible effect of tau pathology in LBD-Ltau cases. Since our study was conducted to mainly examine the effect of α-synuclein pathology, the proportion of subjects having severe tau pathology of Braak NFT stages V–VI was low, less than 10%. The low proportion of this subgroup might therefore have influenced our results. Nevertheless, it is notable that tau burden in the hippocampal dentate gyrus tended to be more severe in TDP-43-positive LBD cases, and that tau was often colocalized with TDP-43 in the amygdala. The independent effects on the development of TDP-43 accumulation of tau pathology, as well as that of α-synuclein pathology, need to be further examined in future studies using a multivariate model in a larger number of cases with various degrees of tau and α-synuclein pathologies.
The pathophysiological mechanism underlying the coexistence of α-synuclein and TDP-43 accumulations in the same LBD case remains unclear. It has been reported that some FTLD-TDP cases with progranulin gene mutations had concomitant α-synuclein pathology [5, 24], although the frequency is not high: in a previous study, only one of 18 cases (5.5%) of FTLD with ubiquitin-positive inclusion (FTLD-U) had Lewy pathology [18]. It was also reported that one case of familial PD (α-synuclein A53T heterozygote) had TDP-43 pathology [26]. To our knowledge, the frequency of TDP-43 pathology in familial LBD cases has not been examined. Colocalization of TDP-43 and α-synuclein in DLB cases was demonstrated in two studies [2, 14], being consistent with our findings. An ultrastructural study also demonstrated that filaments and granular material associated with α-synuclein filaments in Lewy bodies were labeled with anti-TDP-43 antibodies [25].
It is difficult to draw any definite conclusions regarding the biological mechanism underlying the coexistence of α-synuclein and TDP-43 in the same neuron or the same case. However, the results presented in this paper suggest that the limbic system, and in particular the amygdala, is vulnerable to the deposition of TDP-43 in LBD, as well as other degenerative diseases including AD [1, 2, 15], AGD [8], and PSP [34]. Therefore, TDP-43 deposition in the amygdala may be a region-specific rather than disease-specific phenomenon. In LBD, α-synuclein deposition in the limbic region may be primary, and TDP-43 secondarily deposits upon pre-existing LBs, generating some colocalization. This hypothesis seems to be supported by observations that some TDP-43-positive inclusions in our cases showed typical morphological features of LBs. However, the existence of TDP-43 deposited separately from α-synuclein accumulation may also suggest that TDP-43 accumulation cannot only be explained by some direct biological synergy between the proteins. Further, severe neurodegeneration associated with α-synuclein deposition might indirectly lead TDP-43 to accumulate in the vulnerable regions, in particular the amygdala. In addition, the possibility that the accumulation of TDP-43 might be associated with aging should not be excluded. Indeed, some previous studies have demonstrated that aging influences the accumulation of TDP-43 in AD [2, 20] and PDD [28], with age at death being later in cases with TDP-43 changes than in those without. Recently, Geser et al. [10] proposed that TDP-43 proteinopathies could be divided into two categories, major TDP-43 proteinopathies (e.g., ALS and FTLD-TDP [3, 6, 7, 29]) and disorders with secondary TDP-43 pathologies (e.g., AD [1, 2, 15], LBD [2, 14, 28], AGD [8], CBD [32], and PSP [34]). Potential mechanisms regarding TDP-43 accumulation (as mentioned above) could be associated with the pathophysiology in the latter category.
TDP-43 pathology is known to be strongly associated with the development of hippocampal sclerosis. For example, it was reported that 59 of 75 cases (79%) of FTLD with ubiquitin-positive inclusions (FTLD-U) had hippocampal sclerosis [19], 8 of 11 cases (73%) of pure hippocampal sclerosis had TDP-43-positive inclusions [1]. With respect to neuronal loss in the hippocampus in LBD cases with TDP-43 pathology, Nakashima-Yasuda et al. [28] reported that the frequency of hippocampal sclerosis was 60% of 25 TDP-43-positive DLB + AD cases, and 50% of 4 TDP-43-positive PDD cases, and none of 5 TDP-43-positive PD cases. Why none of the present LBD cases with TDP-43 pathology had hippocampal sclerosis is unclear. However, one plausible explanation is that TDP-43 pathology in our LBD series might less severe than that in LBD series by Nakashima-Yasuda: all of their TDP-43-positive LBD cases had the labeled inclusions in the hippocampal dentate gyrus, while only 30% of TDP-43-positive cases in our series had the lesions in the site. As with the relationship with hippocampal sclerosis, it is also plausible that the severity of TDP-43 pathology might have an impact on clinical presentation in LBD cases. However, the fact that no clear association between TDP-43 pathology and presence or absence of dementia was noted in our LBD cases might be explained by the relatively mild TDP-43 pathology in our series. The relationship between the severity of TDP-43 pathology and the development of hippocampal sclerosis, and the effects of these pathological parameters on clinical presentation in LBD cases should be examined in the future.
References
Amador-Ortiz C, Lin WL, Ahmed Z et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445
Arai T, Mackenzie IR, Hasegawa M et al (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Cairns NJ, Neumann M, Bigio EH et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22
Davidson Y, Kelley T, Mackenzie IR et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113:521–533
Fujishiro H, Uchikado H, Arai T et al (2009) Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 117:151–158
Geser F, Winton MJ, Kwong LK et al (2008) Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115:133–145
Geser F, Martinez-Lage M, Kwong LK et al (2009) Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol 256:1205–1214
Hansen L, Salmon D, Galasko D et al (1990) The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology 40:1–8
Hasegawa M, Arai T, Akiyama H et al (2007) TDP-43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 130:1386–1394
Hasegawa M, Arai T, Nonaka T et al (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70
Higashi S, Iseki E, Yamamoto R et al (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184:284–294
Hu WT, Josephs KA, Knopman DS et al (2008) Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 116:215–220
Inukai Y, Nonaka T, Arai T et al (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett 582:2899–2904
Jellinger KA (2010) The neuropathologic substrate of Parkinson disease dementia. Acta Neuropathol 119:151–153
Josephs KA, Ahmed Z, Katsuse O et al (2007) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 66:142–151
Josephs KA, Dickson DW (2007) Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging 28:1718–1722
Josephs KA, Whitwell JL, Knopman DS et al (2008) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70:1850–1857
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) Striatal beta-amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol 67:155–161
Kalaitzakis ME, Pearce RK (2009) The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol 118:587–598
Kosaka K (1990) Diffuse Lewy body disease in Japan. J Neurol 237:197–204
Leverenz JB, Yu CE, Montine TJ (2007) A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain 130:1360–1374
Lin WL, Dickson DW (2008) Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 116:205–213
Markopoulou K, Dickson DW, McComb RD (2008) Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Variability in familial Parkinson’s disease. Acta Neuropathol 116:25–35
McKeith IG, Dickson DW, Lowe J et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
Nakashima-Yasuda H, Uryu K et al (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114:221–229
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Obi K, Akiyama H, Kondo H et al (2008) Relationship of phosphorylated alpha-synuclein and tau accumulation to Abeta deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol 210:409–420
The National Institute on Aging, and Reagan Institute Working Group (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease. The National Institute on Aging, and Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. Neurobiol Aging 18:S1–S2
Uryu K, Nakashima-Yasuda H, Forman MS et al (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564
Yokota O, Tsuchiya K, Uchihara T et al (2007) Lewy body variant of Alzheimer’s disease or cerebral type Lewy body disease? Two autopsy cases of presenile onset with minimal involvement of the brainstem. Neuropathology 27:21–35
Yokota O, Davidson Y, Bigio EH et al (2010) Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 120:55–66
Acknowledgments
We would like to thank Dr. F. Higaki (Department of Radiology, Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences) for the statistical analysis, Ms. M. Onbe (Department of Neuropsychiatry. Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences) for their excellent technical assistance. We also acknowledge and thank the Parkinson’s Disease Society UK Brain Bank for making available tissue samples for this study. This study was supported in part by a research grant from the Uehara Memorial Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yokota, O., Davidson, Y., Arai, T. et al. Effect of topographical distribution of α-synuclein pathology on TDP-43 accumulation in Lewy body disease. Acta Neuropathol 120, 789–801 (2010). https://doi.org/10.1007/s00401-010-0731-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0731-9