
Abstract
Dynamic viscoelasticity measurements were carried out for concentrated solutions of linear d-glucans in BmimCl to examine the effect of the linkage between repeating units of glucose on the rheological properties. The values of molecular weight between entanglements (M e) were determined for four d-glucans: curdlan, pullulan, cellulose, and amylose. From the concentration dependence of M e, the value of M e in the molten state (M e,melt) for each d-glucan was estimated as a material constant. The order of M e,melt became cellulose < pullulan < curdlan < amylose, indicating that the linkage is actually influential in M e,melt for the linear d-glucans. The relationship between M e,melt and the molecular structure of the d-glucans were discussed assuming that the values of M e,melt for the d-glucans primarily reflect the chain stiffness such as the characteristic ratio C ∞ on the analogy of synthetic polymers. Although the trend was not so clear, it was shown that N unit is a decreasing function of C ∞ .
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The molecular weight between entanglements (M e) is a material constant for polymers to characterize the rheological properties at long times [1, 2]. So far, M e has been obtained for various kinds of melts of synthetic polymers [1], but the relationship between M e and the molecular structure of the polymers has not yet been fully understood [3–5]. Concerning polysaccharides, because M e values available are quite limited [6–10], there is little information on the relationship with the molecular structure. We have reported [10] that M e for galactomannans, which commonly have a side group of galactose, is not so sensitive to the galactose/mannose ratio, and therefore, the different introduction ratio of the bulky side group is not so appreciable for the polysaccharides. The average spacing of entanglement networks for linear polysaccharides, such as cellulose, agarose, and gellan, appeared to be almost independent of species of the repeating unit (linear polysaccharides here mean polysaccharides without a side group of monosaccharide or larger), but amylose had much larger M e than the other polysaccharides probably due to a helical nature of amylose chain in a local scale [7]. Amylose and cellulose are categorized in linear d-glucans, and the linear d-glucans have a wide variety of molecular structures depending on the linkage between repeating units of d-glucose. Above-mentioned results suggests that the linkage type is influential in M e and therefore in the rheological properties of the linear d-glucans. Systematic studies for d-glucans will give us more detailed information on the effect of the linkage type between repeating units on M e. It should be also noted that such a variation based on the difference in linkage is characteristic of polysaccharides.
In this study, rheological properties were examined systematically for four kinds of linear d-glucans: pullulan, curdlan, cellulose, and amylose. Although rheological studies of the latter two polysaccharides were performed previously [7], we were able to obtain another grade of the polysaccharides and therefore tried to check the reproducibility for the polymers. The linkage of d-glucose units is β-(1,3) bond for curdlan; the sequence of α-(1,4), α-(1,4), and α-(1,6) bonds for pullulan [11]; β-(1,4) bond for cellulose; and α-(1,4) bond for amylose. Concentrated solutions of these glucans were prepared using an ionic liquid as a solvent and dynamic viscoelasticity was measured for the solutions. The values of M e in the molten state (denoted by M e,melt in the following part) for the glucans were estimated from the concentration dependence of M e in the solutions. To our knowledge M e,melt for curdlan and pullulan were firstly determined in this study, while M e,melt for cellulose and amylose were reevaluated using samples from a different manufacturer.
Experimental
Materials
Four linear d-glucans, curdlan (Wako, Japan), pullulan (Wako, Japan), cellulose (Aldrich, USA), and amylose (Aldrich, USA) were used without further purification. The curdlan sample was ground with a pestle into fine grains prior to the solution preparation. The weight average molecular weight (M w; in grams per mole) of the pullulan sample was reported to be from 5 × 104 through 1 × 105 by the supplier, whereas M w of the rest of samples were unavailable. An ionic liquid of 1-butyl-3-methylimidazolium chloride (BmimCl; Aldrich, USA) was used without further purification as a solvent. According to the manufacturer’s data sheet, the melting point of BmimCl was reported to be 70 °C. The BmimCl solutions of the d-glucans were prepared as follows. The powdery samples of the glucans were added into preheated BmimCl in a dry glass vessel, and the mixture was quickly stirred with a stainless spatula on a hot plate at 80 °C. The mixtures were kept on a hot plate at 80 °C for 1 day for curdlan and cellulose and at 80 °C for 1 day and then at 100 °C for 1 day for pullulan. For amylase, the mixture was put in a vacuum oven at 120 °C and then kept for more than 12 h. The concentration of the d-glucans (c) ranged from 1.4 × 102 to 2.7 × 102 kg m−3 (about 13–25 wt%) for curdlan, from 4.2 × 102 to 5.2 × 102 kg m−3 (about 40–50 wt%) for pullulan, from 5.4 × 101 to 2.1 × 102 kg m−3 (about 5–20 wt%) for cellulose, and from 4.2 × 102 to 5.2 × 102 kg m−3 (about 40–50 wt%) for amylose. In the calculation of c, the density of BmimCl was assumed to be 1.08 × 103 kg m−3, as in the case of previous studies [7–10]. The densities for the d-glucans were commonly assumed to be 1.0 × 103 kg m−3, since the values for purely amorphous d-glucans are not available at present [7]. The sample preparation was carried out just before rheological measurements.
Measurements
Dynamic viscoelasticity measurements were carried out with a rheometer (ARES, now TA Instruments, USA) under nitrogen atmosphere. A cone-plate geometry with the diameter of 25 mm and the cone angle of 0.1 rad was used. The temperature (T) ranged from 20 to 120 °C. Because the supercooled solutions were rather stable, we were able to execute the viscoelasticity measurements for the d-glucan solutions below the melting temperature of BmimCl (even at 20 °C) [6–10]. The angular frequency (ω) dependence curves of the storage modulus (G′) and the loss modulus (G″) were measured in the range of ω from 0.01 to 100 s−1. The amplitude of the oscillatory strain (γ) was set at 0.1 so that the linear viscoelasticity was realized; in some cases, however, a smaller γ of 0.01 was chosen to avoid slippage between the sample and the metal surface of the fixture. Finally, the master curves of G′ and G″ were prepared by applying the time (or angular frequency)–temperature superposition principle to the G′ and G″ curves at individual temperatures. The reference temperature (T r) was set to be 80 °C, and the shift factor in the horizontal direction was designated by a T.
Results and discussion
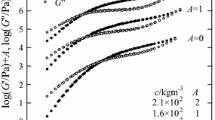
Figure 1 shows the master curves of G′ and G″ for the curdlan solutions. To avoid overlapping, the curves are shifted upwards by a factor A as c increases. As can be seen from the figure, the applicability of the time–temperature superposition principle is fine for four solutions shown here. The shift factor a T at a given temperature was almost independent of c although the data are not shown here, and the temperature dependence of a T were well represented by an Arrhenius-type equation. On each G′ curve, the so-called rubbery plateau is observed and the plateau becomes wider as c increases, although the plateau has a tilt due to a broad molecular weight distribution of curdlan used. The emergence of the plateau means that the entanglement coupling exists at any concentration shown in the figure. For solutions at c = 1.4 × 102 and 1.6 × 102 kg m−3, the flow region can also be seen at low ωa T. The master curves of G′ and G″ for the pullulan solutions at T r = 80 °C are shown in Fig. 2. Here, the vertical shift by A is made for the master curves, as is the case of Fig. 1. The rubbery plateau appears on each curve, although the plateau for pullulan is not as clear as for curdlan shown in Fig. 1. It should be noted for pullulan used here that the solutions at c < 4.2 × 102 kg m−3 showed no rubbery plateau or, in other words, no entanglement coupling. In the figure, the flow zone is clearly observed at low ωa T for each pullulan solution. Figure 3 shows the similar plots for the cellulose solutions. The rubbery plateau can be clearly observed in the middle ωa T region. The master curves for the amylose solutions are shown in Fig. 4. The master curves for amylose resemble those for pullulan in shape, but the rubbery plateau as well as the flow zone appears for all solutions.

Master curves of ω dependence of G′ and G′′ for curdlan solutions at T r = 80 °C. The curves are shifted upwards by A

Master curves of ω dependence of G′ and G′′ for pullulan solutions at T r = 80 °C. The curves are shifted upwards by A

Master curves of ω dependence of G′ and G′′ for cellulose solutions at T r = 80 °C. The curves are shifted upwards by A

Master curves of ω dependence of G′ and G′′ for amylose solutions at T r = 80 °C. The curves are shifted upwards by A
From the analogy with the rubber elasticity, M e at a polymer concentration of c can be given by
Here, \( G_N^0 \) is the plateau modulus, and R is the gas constant [1, 2, 12]. As stated previously, the G′ curves obtained in this study were tilted, and therefore, we defined \( G_N^0 \) as the G′ value at ωa T where the loss tangent (tan δ = G′′/G′) vs. ωa T curve attained the minimum in the rubbery region [6–10]. For example, \( G_N^0 \) for the curdlan solution at c = 2.7 × 102 kg m−3 is determined to be 1.9 × 104 Pa, which gives M e of 4.1 × 104 from Eq. 1 with T = T r. The values of M e for the other solutions in Figs. 1, 2, 3, and 4 can be obtained in a similar manner. Now that we have the information of M w for pullulan (M w ranging from 5 × 104 to 1 × 105), we can check the reliability of obtained M e for pullulan by comparing the value with M w. Generally M e is c-dependent, and the largest M e is realized at the critical concentration for entanglements below which the entanglements vanish. For the largest value of M e, we can expect M w = 2M e [1]. For pullulan, the solution at c = 4.2 × 102 kg m−3 showed the rubbery plateau (which gives M e of 2.6 × 104), but at lower c, we did not have the plateau. This suggests that c of 4.2 × 102 kg m−3 is close to the critical concentration. If we take c of 4.2 × 102 kg m−3 as the critical one, then we have 5.2 × 104 for M w from M w = 2M e. This value is close to the lower bound of the supplier’s data of 5 × 104–1 × 105. The rubbery plateau was not so clear for pullulan (Fig. 2), but the above discussion strongly supports the entanglements really exist at c higher than the critical concentration.
In Fig. 5, the values of log M e for the liner d-glucans are plotted against log c. This figure also contains the results for the cellulose and amylose obtained in the previous study [7]. As no marked difference (or splitting) appears to exist between the previous and current data for the polymers, we can conclude for the two series of polymers that M e data are reproducible. A straight line with a slope of −1 is drawn to fit the plots for each d-glucan since we have the knowledge that M e is inversely proportional to c for many polymer systems [2, 13, 14]. It is seen that data points for each d-glucan are well fitted by the straight line, indicating that the c −1 dependence of M e really holds for the d-glucans in this study. Assuming the density of all d-glucans to be 1.0 × 103 kg m−3, M e,melt for the d-glucans can be determined as the value of M e at c = 1.0 × 103 kg m−3 in Fig. 5. The obtained values of M e,melt are 1.2 × 104, 1.1 × 104, 3.5 × 103, and 2.4 × 104 for curdlan, pullulan, cellulose, and amylose, respectively. In the previous study, we had M e,melt of 3.2 × 103 for cellulose and that of 2.5 × 104 for amylose [7], which are very close to the current results. It is also possible to use another measure describing the entanglement spacing in melt, namely, the number of monosaccharide units between entanglements in the molten state (N unit). We define N unit by N unit = M e,melt/M unit, with M unit being the molecular weight of the repeating unit. For the linear d-glucans used in this study, M unit is identical for the four polymers (i.e., M unit = 162), so that N unit becomes 71, 68, 22, and 148 for curdlan, pullulan, cellulose, and amylose, respectively. These are summarized in Table 1 together with M e,melt. We do not know exactly why M e,melt (or equivalently N unit) differs from a d-glucan to another, but this fact strongly suggests that some molecular parameters such as chain stiffness affect the entanglement spacing characterized by M e,melt. In the previous study [7], we found a considerable difference in M e,melt between cellulose and amylose, and the difference was attributed to a helical nature of amylose chain in a local scale although amylose chain has a random coil conformation as cellulose chain has in a global scale. For pullulan and curdlan, it have been reported that the local conformation of a curdlan chain is close to the 6/1 helix like an amylose chain [15], while pullulan has no helical nature [16].

Double-logarithmic plot of M e against c for d-glucans in solution. Each line is the best fit one with a slope of −1. M e,melt for d-glucans is determined as M e at c = 103 kg m−3. Data for cellulose (plus symbol) and amylose (ex symbol) in the previous study7 are added
Several attempts have been made to connect M e,melt with the characteristic ratio C ∞, a well-known molecular parameter of real (flexible) polymer chain. For example, Graessley and Edwards have considered that \( G_N^0 \) is a function of the contour chain length per unit volume as well as of the Kuhn step length (=C ∞ l 0; l 0 being the average length of the main chain bonds) [3], which gives M e,melt ∝ C −1∞ if parameters other than M e,melt and C ∞ are eliminated. Wu [4] has obtained another expression of M e,melt ∝ C 2∞ by using a pseudo-topological model. Fetters et al. [5] have proposed M e,melt ∝ C −3∞ if we focus on just M e,melt and C ∞. Although the relation between M e,melt and C ∞ is still a matter of controversy even for synthetic polymers, the relationship between M e,melt and C ∞ is now considered for the linear d-glucans. Of course, we encounter another problem in this case that we do not have C ∞ from a single data source, but the fact that C ∞ has been reported implies for these glucans in solution that the favorable chain conformation is a random coil. Even at high c where entanglements are generated, the random-coil conformation appears to be favorable for glucans. In Table 1, C ∞ for the d-glucans is also listed with the source. Quoted values are all experimental data in the literature, although the measurement conditions are not necessarily the same [11, 16–26]. The data of C ∞ obtained by model calculations were not employed because they have large scattering from researcher to researcher. Figure 6 shows the N unit vs. C ∞ plots for the linear d-glucans, where each bar stands for a pair of N unit and C ∞ range. We cannot find out a clear trend from the figure, but it may appear that N unit is a decreasing function of C ∞. In this case, the Wu relation should be excluded for the linear d-glucans.

N unit vs. C ∞ plots for four d-glucans. The range of C ∞ plots for each d-glucan is indicated by a horizontal bar
Conclusions
The values of M e,melt as well as N unit for linear d-glucans were determined from the dynamic viscoelasticity measurement for their concentrated solution in BmimCl. It was found that M e,melt as well as N unit for linear d-glucans depends much on the linkage between glucose repeating units. The order of M e,melt was determined to be cellulose < pullulan < curdlan < amylose. The relationship between M e,melt and the molecular structure of the d-glucans was considered assuming that M e,melt can be connected with the chain stiffness C ∞, as attempted for synthetic polymers. A subtle trend that N unit, namely, M e,melt, is a decreasing function of C ∞ was obtained.
References
Ferry JD (1980) Viscoelastic properties of polymers. Wiley, New York
Doi M, Edwards SF (1986) The theory of polymer dynamics. Clarendon, Oxford
Graessley WW, Edwards SF (1981) Entanglement interactions in polymers and the chain contour concentration. Polymer 22:1329–1334
Wu S (1989) Chain structure and entanglement. J Polym Sci B Polym Phys 27:723–741
Fetters LJ, Lohse DJ, Richter D, Witten TA, Zirkel A (1994) Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 27:4639–4647
Horinaka J, Honda S, Takigawa T (2009) Rheological properties of concentrated solutions of gellan in an ionic liquid. Carbohydr Polym 78:576–580
Horinaka J, Yasuda R, Takigawa T (2011) Entanglement properties of cellulose and amylose in an ionic liquid. J Polym Sci B Polym Phys 49:961–965
Horinaka J, Yasuda R, Takigawa T (2011) Entanglement network of agarose in various solvents. Polymer J 43:1000–1002
Horinaka J, Yasuda R, Takigawa T (2012) Rheological properties of concentrated solutions of agarose in ionic liquid. J Appl Polym Sci 123:3023–3027
Horinaka J, Yasuda R, Takigawa T (2012) Rheological properties of concentrated solutions of galactomannans in an ionic liquid. Carbohyr Polym 89:1018–1021
Buliga GS, Brant DA (1987) Temperature and molecular weight dependence of the unperturbed dimensions of aqueous pullulan. Int J Biol Macromol 9:71–76
Onogi S, Masuda T, Kitagawa K (1970) Rheological properties of anionic polystyrenes. I. dynamic viscoelasticity of narrow-distribution polystyrenes. Macromolecules 3:109–116
Masuda T, Toda N, Aoto Y, Onogi S (1972) Viscoelastic properties of concentrated solutions of poly(methyl methacrylate) in diethyl phthalate. Polym J 3:315–321
Nemoto N, Ogawa T, Odani H, Kurata M (1972) Shear creep studies of narrow-distribution poly(cis-isoprene). III. concentrated solutions. Macromolecules 5:641–644
Rees DA, Scott WE (1971) Polysaccharide conformation. Part VI. Computer model-building for linear and branched pyranoglycans. Correlations with biological function. Preliminary assessment of inter-residue forces in aqueous solution. Further interpretation of optical rotation in terms of chain conformation. J Chem Soc B 1971:469–479
Kato T, Okamoto T, Tokuda T (1982) Solution properties and chain flexibility of pullulan in aqueous solution. Biopolymers 21:1623–1633
Nakata M, Kawaguchi T, Kodama Y, Konno A (1998) Characterization of curdlan in aqueous sodium hydroxide. Polymer 39:1475–1481
Hirano I, Einaga Y, Fujita H (1979) Curdlan (bacterial β-1,3-glucan) in an cadoxen–water mixture. Polym J 11:901–904
Zhang H, Nishinari K (2009) Characterization of the conformation and comparison of shear and extensional properties of curdlan in DMSO. Food Hydrocolloids 23:1570–1578
Brant DA, Burton BA (1981) Solution properties of polysaccharides, Chap 7. American Chemical Society, Washington, pp 81–99
Saalwaechter K, Burchard W, Kluefers P, Kettenbach G, Mayer P, Klemm D, Dugarmaa S (2000) Cellulose solutions in water containing metal complexes. Macromolecules 33:4094–4107
Cai J, Liu Y, Zhang L (2006) Dilute solution properties of cellulose in LiOH/urea aqueous system. J Polym Sci B Polym Phys 44:3093–3101
Nakanishi Y, Norisuye T, Teramoto A, Kitamura S (1993) Conformation of amylose in dimethyl sulfoxide. Macromolecules 26:4220–4225
Norisuye T (1994) Viscosity behavior and conformation of amylose in various solvents. Polym J 26:1303–1307
Goebel KD, Brant DA (1970) The configuration of amylose and its derivatives in aqueous solution. Experimental results. Macromolecules 3:634–643
Ring SG, I’Anson KJ, Morris VJ (1985) Static and dynamic light scattering studies of amylose solutions. Macromolecules 18:182–188
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Horinaka, Ji., Okuda, A., Yasuda, R. et al. Molecular weight between entanglements for linear d-glucans. Colloid Polym Sci 290, 1793–1797 (2012). https://doi.org/10.1007/s00396-012-2728-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-012-2728-5