
Abstract
Receptor for advanced glycation end products (RAGE) is associated with inflammation and the progression of cardiovascular diseases. The current study tested the hypothesis that RAGE is involved in the pathogenesis of aortic valve (AV) calcification. Pioglitazone attenuated AV calcification in experimental hypercholesterolemic rabbits via down-regulation of RAGE. Male New Zealand rabbits weighing 2.5–3.0 kg were randomly divided into three groups: control group, high cholesterol + vitamin D2 (HC + vitD2) group and HC + vitD2 supplemented with pioglitazone group. Compared with HC + vitD2 group, pioglitazone significantly inhibited the progression of AV calcification assessed by echocardiography. HC + vitD2 diet markedly increased RAGE expression, oxidative stress, inflammatory cells infiltration and osteopontin expression. These changes were also significantly attenuated by administration of pioglitazone. Cultured porcine aortic valve interstitial cells (VICs) were used as in vitro model. We found that advanced glycation end products of bovine serum albumin markedly increased the expression of RAGE, induced high levels of production of pro-inflammatory cytokines and promoted osteoblastic differentiation of VICs. However, these effects were found to be remarkably suppressed by siRNA silencing of RAGE and pioglitazone as well. Our data provide evidence that RAGE activation-induced inflammation promotes AV calcification in hypercholesterolemic rabbits, which can be attenuated by pioglitazone treatment. This beneficial effect is associated with remarkable down-regulation of RAGE expression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Aortic valve (AV) calcification is a common clinical problem with increased morbidity and mortality: approximately 2.8 % of adults over 75 years old have some degree of AV calcification [5, 34], and up to 25 % of adults over 65 have valvular sclerosis [3]. Currently, treatment strategies for symptomatic AV calcification only include careful monitoring and judicious timing of AV replacement, yet not everyone is fit or well tolerated for surgery.
AV calcification has traditionally been considered as a degenerative disorder. It involves passive deposition of calcium on AV leaflets. Recently, studies have demonstrated that AV calcification has many features characteristic of an active pathobiological process akin to atherosclerosis, including chronic inflammation, endothelial dysfunction, lipoprotein deposition and extracellular matrix remodeling [36, 37]. Despite these considerable advances, the mechanisms for AV calcification remain to be elucidated.
Receptor for advanced glycation end products (RAGE) is a multi-ligand receptor expressed in a range of cell types, including endothelial, smooth muscle, mesangial and mononuclear cells [6, 33]. The associations among RAGE, inflammation and arterial calcification have been well documented [8, 16, 27]. However, the potential role of RAGE activation in AV calcification has not yet been investigated. Previous studies have shown that metabolic syndrome and diabetes are associated with increased risks of the development of AV calcification [24], and these patients demonstrate significantly increased levels of advanced glycation end products (AGEs) in plasma and tissues. In addition, circulating soluble RAGEs, the extracellular ligand binding domain of RAGE, which prevent AGEs from binding to tissue RAGEs, were found to be significantly lower in the plasma of patients with AV calcification [2]. All these observations imply that RAGE activation may participate in the pathogenesis of AV calcification.
Peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor, is highly expressed in all cells involved in vascular pathologies, including macrophages, endothelial cells, and smooth muscle cells (SMCs) [17]. PPARγ agonists possess anti-inflammatory and anti-oxidant properties [40]. Accumulating data indicate that PPARγ agonists can down-regulate the expression of RAGE both in vitro and in vivo [18, 49]. However, as far as we know, it remains unclear whether PPARγ agonists can protect against the progression of AV calcification.
In this study, we investigated whether RAGE activation is involved in the pathogenesis of AV calcification by promoting inflammatory pathways; and whether pioglitazone, a predominant PPARγ agonist, can alleviate the progression of AV calcification by down-regulating RAGE and inhibiting RAGE-mediated osteoblastic differentiation of valve interstitial cells (VICs).
Materials and methods
Experimental rabbit model
The Institutional Animal Research Committee of Tongji Medical College approved the study protocol, and all animal care conformed to the Guide for the Care and Use of Laboratory Animals. Twenty male New Zealand rabbits (weighing 2.5–3.0 kg) were randomly divided into three groups: (1) Control group (Control; n = 6), rabbits were fed normal rabbit chow without any dietary supplement; (2) High cholesterol + vitamin D2 group (HC + vitD2, n = 7), rabbits were fed 0.5 % cholesterol-enriched chow plus 50,000 IU/day vitamin D2 in drinking water; (3) Pioglitazone treatment group (HC + vitD2 + Pio, n = 7), some HC + vitD2 rabbits concomitantly received 3 mg/kg pioglitazone daily by oral gavage. All animals were housed at room temperature with a 12-h light–dark cycle and fed ad libitum for 12 weeks.
Echocardiography and tissue processing
Transthoracic echocardiography was performed at baseline and at the end of a 12-week intervention. Animals were sedated with intramuscular injection of ketamine (30 mg/kg) and xylazine (3.5 mg/kg). The anterior thorax was shaved for better echo imaging. Images were acquired with a 10 MHZ phased-array probe connected to a GE Vivid 7 ultrasound system (GE Healthcare, UK). Echocardiographic data and related calculations were performed by an experienced operator blind to the assignments as previously described [25]. Aortic valve area (AVA) index was calculated by AVA divided by total body surface area.
After the final echocardiography examination, rabbits were euthanized by a lethal dose of penthobarbital sodium. Immediately after removal from the heart, AV were fixed in 4 % paraformaldehyde for 24 h and then embedded in paraffin for morphological and immunohistochemical examinations.
Cell culture and treatment
Porcine aortic VICs were isolated from AV leaflets of pigs from a local abattoir using collagenase digestion as previously described [19, 42]. Briefly, isolated leaflets were digested in DMEM (Gibco, Carlsbad, CA, USA) containing 1 mg/ml collagenase type II (Invitrogen, Carlsbad, CA, USA) at 37 °C for 30 min. After removal of endothelial cells by vortexing, the leaflets were further digested with a fresh solution of 1 mg/ml collagenase medium for 4–6 h at 37 °C. After vortexing and repeated aspirating to break up the tissue mass, the suspension was spun at 1,000 rpm for 10 min to precipitate cells. Cells were resuspended and cultured in DMEM, supplemented with 100 mg/ml penicillin, 100 U/ml streptomycin, and 10 % FBS (Gibco) in an incubator with 5 % CO2 at 37 °C. To characterize VICs, immunofluorescent staining was applied with antibodies against α-SMA (1:50 dilution, Sigma-Aldrich, St. Louis, MO, USA) and vimentin (1:50 dilution, Boster, China) (Supplemental Fig. 1). All experiments were performed with cells in passages 2–5. For all experiments, cells were grown to 70–80 % confluence, serum-starved overnight, and then incubated with indicated concentrations of AGE-modified bovine serum albumin (AGE-BSA, Biovision, Milpitas, CA, USA), pioglitazone (Sigma-Aldrich) or Bay 11-7082 (Merck, Germany) for indicated periods of time in the presence of 10 mM β-glycerophosphate (Sigma-Aldrich), 100 nM dexamethasone (Sigma-Aldrich), and 50 μg/ml ascorbic acid (Sigma-Aldrich).
siRNA knock-down of RAGE expression
A pool of three target-specific 19-nt siRNA designed to knock down Sus scrofa RAGE (NM_001123218.1) was synthesized and purified by RiboBio (China). Scramble siRNA duplexes with non-specific sequences served as negative controls. Then 30–50 % confluent VICs were transfected with 50 nM siRNA using Lipofectamine 2000 (Invitrogen) and Opti-MEM (Gibco) according to the manufacturer’s recommendations. The medium was changed 6 h after transfection. 72 h later, cells were harvested for analysis of protein expression.
Real-time polymerase chain reaction (PCR) RNA analysis
Real-time PCR was used to quantify the expression of mRNAs encoding interleukin (IL)-6, IL-8, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1) with expression of β-actin as endogenous control. RNA was isolated and reverse transcribed to cDNA as previously described [7]. Real-time PCR assays were carried out using a SYBR®Premix Ex Taq™ (Takara) on a StepOnePlus™ Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Primers used specially for porcine samples were as follows: IL-6 (F: 5′-ATC AGG AGA CCT GCT TGA TG-3′, R: 5′-TGG TGG CTT TGT CTG GAT TC-3′), IL-8 (F: 5′-TCC TGC TTT CTG CAG CTC TC-3′, R: 5′-GGG TGG AAA GGT GTG GAA TG-3′), TNF-α (F: 5′-CCA ATG GCA GAG TGG GTA TG-3′, R: 5′-TGA AGA GGA CCT GGG AGT AG-3′), MCP-1 (F: 5′-GTC ACC AGC AGC AAG TGT C-3′, R: 5′-CCA GGT GGC TTA TGG AGT C-3′), β-actin (F: 5′-GAC CTG ACC GAC TAC CTC-3′, R: 5′-GCT TCT CCT TGA TGT CCC-3′). The PCR conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 40 cycles with melting curve analysis at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. Data were analyzed using a comparative \( 2^{{ - \Updelta \Updelta CT }} \) method.
Western blotting analysis
Cells were harvested and homogenized in RIPA Lysis and Extraction Buffer with protease and phosphatase inhibitor cocktails (Pierce, Rockford, IL, USA). Cytosolic and nuclear proteins were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce). All these procedures were performed according to the manufacturer’s protocols. Equal amounts of protein were resolved by SDS-PAGE (10 % resolving gel with 4 % stacking) and transferred to PVDF membranes. Membranes were blocked with buffer containing 10 % non-fat milk and 5 % BSA in TBS-T (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 0.1 % Tween-20), and then incubated with the following antibodies: RAGE (1:2,000 dilution), bone morphogenetic protein 2 (BMP2, 1:2,000 dilution), osteopontin (1:2,000 dilution), IκBα (1:2,000 dilution), and NF-κB p65 (1:2,000 dilution) from Bioworld Technology, Atlanta, GA, USA; runt-related transcription factor 2 (Runx2, 1:2,500 dilution), alkaline phosphatase (ALP, 1:2,000 dilution), lamin B1 (1:1,000 dilution) and β-actin (1:1,000 dilution) from Santa Cruz Biotechnology, Santa Cruz, CA, USA. Blots were washed in TBS-T and incubated with appropriate HRP-conjugated secondary antibodies. The immune complexes were then visualized using ECL reagent (Beyotime, China). Specific bands were quantified by densitometry using Quantity One software (Bio-Rad, Hercules, CA, USA).
Immunocytofluorescence
VICs were seeded in a 96-well plate and treated with AGE-BSA with or without pioglitazone, as described earlier. After treatment, cells were washed with PBS, fixed in 4 % paraformaldehyde and permeabilized with 0.1 % Triton-X 100 for 10 min at room temperature. Non-specific binding was blocked in 10 % goat serum followed by incubation with anti-NF-κB p65 (1:50 dilution, Bioworld Technology) antibody at 4 °C overnight. Cells were washed and then incubated with goat anti-rabbit Cy3-conjugated secondary antibody for 1 h at room temperature. DAPI (Sigma-Aldrich) was used for nuclear counterstaining. Cells were then visualized with an Olympus fluorescence microscope. Image-Pro Plus (Media Cybernetics, Bethesda, MD, USA) was used to merge the images.
Histological and immunohistochemical staining
AVs were cut into 3-μm-thick slices and stained with hematoxylin and eosin (H&E) and with alizarin red. Immunostaining was performed to identify RAGE, α-SMA, macrophage antigen (RAM11), osteopontin, PPARγ, β-catenin, 3-nitrotyrosine and CD3 expression. Briefly, sections were deparaffinized using xylene and graded ethanol washes, and sodium citrate buffer was used for antigen retrieval. After blocking in 10 % goat serum, the following mouse monoclonal antibodies were applied: RAGE (1:100 dilution, Santa Cruz Biotechnology), PPARγ (1:100 dilution, Santa Cruz Biotechnology), α-SMA (1:100 dilution, Sigma-Aldrich), osteopontin (1:50 dilution, Zsbio, China), β-catenin (1:100 dilution, Santa Cruz Biotechnology), 3-nitrotyrosine (1:100 dilution, Santa Cruz Biotechnology), CD3 (1:100 dilution, Santa Cruz Biotechnology) and RAM11 (1:100 dilution, Dako, Carpinteria, CA, USA), overnight at 4 °C. Then, a Two-Step HRP-conjugated anti-mouse kit (Zsbio) was used. Final staining was performed through reaction with DAB (Zsbio) and counterstaining with hematoxylin (Sigma-Aldrich). Image-Pro Plus (Media Cybernetics) was applied to determine quantitative results (positive staining area/total AV area) for two slices of each AV leaflet, and at least two of the three AV leaflets were analyzed per rabbit. The investigators who performed the analysis were blind to the study groups.
Detection of serum cholesterol, LDL, triglyceride, and glucose levels
Rabbits were subject a 12-h fast before collection of blood samples. Serum total cholesterol (TC), LDL, triglyceride (TG), and glucose levels were determined using indicated kits from Biosino, China, according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as mean ± SD. After confirming that all variables were normally distributed using the Kolmogorov–Smirnov test, statistical differences between groups were evaluated by ANOVA followed by Bonferroni’s multiple comparison test. P < 0.05 was accepted as statistically significant.
Results
Development of AV calcification and echocardiographic data in rabbits
Serial echocardiographic examination showed that rabbits fed the cholesterol-enriched diet plus vitD2 developed AV calcification. This is consistent with previous observations [10]. The valves of rabbits in the control group remained normal. Compared with the normal controls, echogenicity was significantly increased in HC + vitD2 rabbits (Fig. 1a, b, d), AVA and AVA index in HC + vitD2-fed rabbits also decreased markedly (Fig. 2a). However, no significant differences in transvalvular systolic velocity were observed among groups (Fig. 2c). Rabbits given pioglitazone daily demonstrated significant reduction in echogenicity (Fig. 1b–d) in AV compared with rabbits in HC + vitD2 group. Pioglitazone treatment also prevented the reduction of AVA index and the difference between final and baseline AVA (Fig. 2b, c).

Echocardiographic data in the rabbit model. a–c Representative echocardiographic images of AV in parasternal short-axis (a1–c1, a2–c2) and long-axis (a3–c3, a4–c4) after 12 weeks of intervention. d Echogenicity enhanced in AV from HC + vitD2 rabbits compared with the control group, and pioglitazone significantly attenuated this effect (Control: n = 6; HC + vitD2: n = 7; HC + vitD2 + Pio: n = 7; **P < 0.01)

Echocardiographic data in the rabbit model. a Absolute AVA after 3 months of intervention: HC + vitD2 diet reduced the absolute value of aortic valve area (AVA) compared with the controls. Pioglitazone treatment attenuated this only slightly. b Differences between final AVA and baseline AVA: increased loss in final AVA versus baseline AVA in HC + vitD2 rabbits compared with the control group, which was significantly protected by pioglitazone administration. c Statistical analysis of AVA index: AVA index reduced significantly in HC + vitD2 treated animals, while pioglitazone prevented this effect. d No significant differences in transvalvular systolic velocity were observed among groups (Control: n = 6; HC + vitD2: n = 7; HC + vitD2 + Pio: n = 7; *P < 0.05 and **P < 0.01)
Previous studies suggested that thiazolidinediones, used for the treatment of patients with type 2 diabetes mellitus (DM2), are associated with an increased incidence of heart failure [15]. However, compared with rosiglitazone, pioglitazone are less prone to induce heart failure [11, 28]. In the present study, there is no evidence that pioglitazone induced heart failure (Supplemental Table 1).
We also compared blood biochemical parameters among groups. After 12 weeks of HC + vitD2 diet, significant increases in serum cholesterol and LDL levels were observed. Pioglitazone treatment did not alter the lipid profiles or glucose levels (Supplemental Table 2).
Altogether, these results suggest that pioglitazone administration exerts a protective effect against the progression of AV calcification. This beneficial effect is independent of changes in serum lipid concentration and in glucose metabolism.
Morphological and immunohistochemical staining
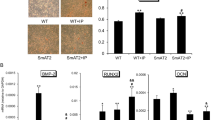
Transition from VICs to myofibroblasts is a critical step in valvular pathology [39]. We examined myofibroblasts using immunohistochemical staining with α-SMA antibody. High-cholesterol diets markedly increased the α-SMA-positive area, but the pioglitazone treatment group showed significantly fewer myofibroblasts in the valve leaflets (Fig. 3).

Representative light microscopy images of rabbit AV. a–c H&E and α-SMA staining of AV in different groups. Scale bar 200 μm, and arrow indicates ×200 magnification. d Quantification data showed that the HC + vitD2 diet markedly increased the number of myofibroblasts in AV compared with the control, but pioglitazone significantly inhibited this effect (Control: n = 6; HC + vitD2: n = 7; HC + vitD2 + Pio: n = 7; **P < 0.01.)
We next evaluated the staining of osteopontin, an important type of bone matrix protein, and found that it presented at high levels in the AV of hypercholesterolemic rabbits. Osteopontin expression was significantly decreased by pioglitazone treatment (Fig. 4a1–c1, d). AV calcification was assessed using alizarin red staining. Calcium deposits in the leaflets were increased in HC + vitD2 group, compared with normal controls. However, only mild calcification was observed when HC + vitD2-fed rabbits were given pioglitazone (Fig. 4a2–c2, e).

Immunohistochemical staining of rabbit AV in different groups. a–c Representative images of immunohistochemical staining of osteopontin (a1–c1) and RAGE (a3–c3); and alizarin red staining (a2–c2) of calcium deposition in AV leaflets. Scale bar 200 μm, and arrow indicates ×200 magnification. Pioglitazone administration notably reduced osteopontin expression (d), calcified area (e), and RAGE expression (f) induced by HC + vitD2 diet (Control: n = 6; HC + vitD2: n = 7; HC + vitD2 + Pio: n = 7; **P < 0.01 and *P < 0.05.)
BMP and Wnt/β-catenin signaling pathways have been reported to be associated with AV calcification [35]. We also found that hypercholesterolemia increased AV β-catenin expression, and that pioglitazone eliminated much of this effect, as shown in Supplemental Fig. 2.
Reactive oxygen species (ROS) were proved to be involved in progression of AV calcification [25]. In HC + vitD2 rabbits, obvious levels of nitro/oxidative stress were observed in AV, as indicated by increased levels of 3-nitrotyrosine immunostaining, which decreased significantly upon pioglitazone administration (Supplemental Fig. 3).
We then examined whether RAGE activation was also induced in the AV calcification model. In accordance with observations made earlier in the project, dietary cholesterol was associated with a significant increase in leaflet RAGE immunostaining, which was dramatically lowered by pioglitazone treatment (Fig. 4a3–c3, f).
Finally, we determined whether hypercholesterolemia or pioglitazone treatment altered PPARγ expression in vivo. No significant change in PPARγ expression was detected after 12 weeks of HC + vitD2 diet induction. As expected, pioglitazone treatment markedly up-regulated AV PPARγ expression in vivo (Supplemental Fig. 4).
Necessity of RAGE for AGE-BSA-induced osteoblastic differentiation of VICs
These findings showed RAGE activation to be involved in AV calcification. This led us to hypothesize that RAGE activation triggers the intracellular signaling that results in osteoblastic differentiation of VICs. To confirm this, we incubated VICs with AGE-BSA (0, 10, 50, and 100 μg/ml), a predominant RAGE ligand, for 72 h. We then examined expression of RAGE and several typical osteoblast phenotypic markers. RAGE and osteopontin levels increased in a dose-dependent manner with increasing concentrations of AGE-BSA (Fig. 5a–c). To explore the mechanism by which RAGE activation leads to the osteoblastic differentiation of VICs, we assessed the expression of BMP2 and Runx2. We observed marked elevation of BMP2 and Runx2 levels after the stimulation (Fig. 5a, c). To validate this, siRNA targeting RAGE was introduced. RAGE siRNA specifically and efficiently knocked down expression of RAGE (Fig. 5d), blunting the up-regulatory effect of AGE-BSA on Runx2, osteopontin (Fig. 5e, f) and ALP expression (Supplemental Fig. 5c). Collectively, our results suggest that activation of RAGE mediates the AGE-BSA-induced osteoblastic differentiation of VICs.

Pioglitazone inhibited RAGE-mediated osteoblastic differentiation of VICs. a Immunoblotting for RAGE, pro-osteogenic signals and osteogenic phenotype markers of VICs exposure to AGE-BSA. AGE-BSA induced RAGE activation (b), BMP2, Runx2 and osteopontin expression (c) in a dose-dependent manner. (n = 3 for each experiment; *P < 0.05 vs. control; **P < 0.01 vs. control). RAGE siRNA significantly reduced RAGE protein (d), and blocked AGE-BSA induced up-regulation of Runx2 and osteopontin (e and f). (n = 3 for each experiment; **P < 0.01 vs. Scramble siRNA; ## P < 0.01 vs. AGE-BSA + Scramble siRNA). Pioglitazone abolished RAGE activation (g and h), and decreased augmentation of Runx2 and osteopontin protein levels induced by AGE-BSA (g and i) (n = 3 for each experiment; **P < 0.01 vs. control; # P < 0.05 vs. AGE-BSA; ## P < 0.01 vs. AGE-BSA)
Effects of pioglitazone on RAGE-mediated osteoblastic differentiation of VICs
Preliminary studies have shown that alleviation of AV calcification and decreased RAGE activation could be achieved by pioglitazone administration in hypercholersterolemic rabbits. We reasoned that pioglitazone was able to prevent RAGE-mediated osteoblastic differentiation of VICs. VICs were pretreated with different concentrations of pioglitazone (50, 100 μM) for 1 h and then stimulated with AGE-BSA (100 μg/ml) for 72 h. Not surprisingly, pioglitazone down-regulated RAGE expression of VICs (Fig. 5g, h), which was in line with in vivo results (Fig. 4a3–c3, f). The levels of expression of pro-osteogenic factors, Runx2, osteopontin, and ALP were also significantly reduced (Fig. 5g, i, Supplemental Fig. 5b).
In vitro effects of AGE-BSA and pioglitazone on VICs inflammatory responses
The critical role of inflammation in the development of AV calcification is widely accepted. RAGE activation leads to persistent inflammation in a variety of cells. Given these findings, we sought to determine whether RAGE activation induces inflammatory response in VICs. We stimulated VICs with AGE-BSA (100 μg/ml) and measured inflammatory cytokine levels 24 h later. Under basal conditions, cultured VICs released low levels of IL-6, IL-8, TNF-α and MCP-1. However, treatment with AGE-BSA significantly augmented the production of IL-8, TNF-α and MCP-1. This augmentation was blocked when RAGE expression was inhibited by siRNA silencing (Fig. 6a–c).

Pioglitazone relieved RAGE-mediated inflammation in VICs. Quantitative PCR analysis of AGE-BSA induced expression of inflammatory gene mRNA in VICs. AGE-BSA markedly augmented mRNA expression of IL-8 (a), TNF-α (b) and MCP-1 (c), and RAGE silencing blocked these effects. (n = 3 for each experiment; **P < 0.01 vs. Scramble siRNA; # P < 0.05 vs. AGE-BSA + Scramble siRNA; ## P < 0.01 vs. AGE-BSA + Scramble siRNA). Pioglitazone pre-incubation significantly decreased expression of IL-8 (d), TNF-α (e) and MCP-1 (f) Mrna stimulated by AGE-BSA (n = 3 for each experiment; **P < 0.01 vs. control; # P < 0.05 vs. AGE-BSA; ## P < 0.01 vs. AGE-BSA)
Next, to determine whether pioglitazone could relieve the inflammation, we pretreated VICs with pioglitazone (50, 100 μM) 1 h before incubation with AGE-BSA. The production of IL-8, TNF-α and MCP-1 decreased (Fig. 6d–f), indicating that pioglitazone inhibited AGE-BSA-induced cellular inflammation.
Pioglitazone inhibits AGE-BSA-induced activation of NF-κB in VICs
To uncover the signal transduction underlying the above observations, we explored whether the NF-κB pathway, the key transcription factor responsible for the production of pro-inflammatory cytokines, was involved in the process. As shown in Fig. 7, stimulating cells with AGE-BSA (100 μg/ml) for 2 h led to significant NF-κB p65 intranuclear translocation (Fig. 7b), as well as increased degradation of IκBα (Fig. 7a). Both effects were inhibited by pioglitazone pretreatment (100 μM). This was further confirmed by Immunocytofluorescence as shown in Fig. 7c–e.

Pioglitazone attenuated AGE-BSA induced NF-κB signaling in VICs. a Pioglitazone prevented AGE-BSA induced degradation of IκBα, as shown by the increase in IκBα levels in cytosolic extract relative to AGE-BSA treatment alone. b Pioglitazone inhibited AGE-BSA induced intranuclear translocation of NF-κB p65, as shown by a decrease in NF-κB p65 levels in nuclear extract relative to AGE-BSA treatment alone. c–e Representative immunocytofluorescence staining of NF-κB p65 in VICs. NF-κB p65 intranuclear translocation was detected in the nuclei of cells exposed to AGE-BSA alone (d1, d3) whereas pioglitazone pretreatment inhibited this translocation (e1, e3) (n = 3 for each experiment; **P < 0.01 vs. control; # P < 0.05 vs. AGE-BSA; ## P < 0.01 vs. AGE-BSA)
We used Bay 11-7082, an inhibitor of NF-κB, to confirm whether NF-κB signaling functionally mediates RAGE-induced osteoblastic differentiation of VICs. In the presence of Bay 11-7082 (10 μM), AGE-BSA-induced elevation of Runx2 and osteopontin was significantly inhibited (Supplemental Fig. 6).
In vivo anti-inflammatory effect of pioglitazone on AV leaflets
After demonstrating the anti-inflammatory effect of pioglitzone by inhibiting RAGE activation, we next evaluated whether pioglitazone exerts protective effects against inflammation in AV leaflets in vivo. Therefore, we identified infiltrating inflammatory cells. HC + vitD2-fed rabbits showed markedly increased macrophages (Fig. 8b, d) and T lymphocytes (Supplemental Fig. 7b, d) infiltration. Concomitant with reduced RAGE activation in AV leaflets (Fig. 4a3–c3, f), pioglitazone treatment greatly attenuated this HC + vitD2-induced effect (Fig. 8c, d, Supplemental Fig. 7c, d). These results suggest that the protective role of pioglitzaone against AV calcification can be probably attributed to its anti-inflammatory effect.

Pioglitazone administration diminished AV macrophages infiltration induced by HC + vitD2 diet in rabbits. RAM11 (macrophage marker) staining in AV leaflets in control group (a), HC + vitD2 group (b) and HC + vitD2 supplemented with pioglitazone group (c). Scale bar 200 μm, and arrow indicates ×200 magnification. d Quantification of RAM11-stained area showed that HC + vitD2 diet significantly increased AV macrophages infiltration compared with control group, but pioglitazone suppressed this (Control: n = 6; HC + vitD2: n = 7; HC + vitD2 + Pio: n = 7; **P < 0.01)
Discussion
The present study shows that oral administration of pioglitazone attenuates the progression of AV calcification in hypercholesterolemic rabbits without significantly altering blood glucose levels. This effect is accompanied by reduced RAGE expression, β-catenin expression, oxidative stress, inflammatory cells infiltration, and osteoblastic markers. Our in vitro studies evaluating the effects of pioglitazone on AGE-BSA stimulated VICs showed that pioglitazone significantly reduced RAGE activation and inhibited NF-κB p65 intranuclear translocation, inflammatory cytokines production, and pro-osteogenic signaling cascades. To our knowledge, the present findings provide the first evidence that pro-inflammatory effects induced by RAGE/ligand interaction in VICs contribute to the development and progression of AV calcification, and that pioglitazone exerts these protective effects against AV calcification by inhibiting RAGE activation and its associated inflammatory responses.
RAGE is a member of the immunoglobulin superfamily which is able to bind to a chemically diverse range of AGEs and other non-AGE ligands, including high-mobility group box-1 protein, S100/calgranulins and β-sheet fibrils [47, 48]. Previous studies have shown that S100A12 transgenic Apoe-Null mice has increased vascular calcification, and S100A12 has been shown to promote osteoblastic gene BMP2, dentin matrix acidic phosphoprotein 1 (Dmp1) and Runx2 expression in vascular SMCs when exposed to an inflammatory environment. In contrast, pretreatment with soluble RAGE, which prevents activation of cell surface receptors such as RAGE, significantly attenuated the expression of osteoblastic genes [16]. Activation of RAGE by AGEs and adenovirus transfection also induced osteoblastic differentiation of vascular SMCs [43, 46]. We therefore explored whether VICs transformed into osteoblast-like cells in response to RAGE activation induced by AGE-BSA. We assessed the expression of BMP2 and Runx2 in cultured VICs after stimulation with AGE-BSA. BMP2 is a potent pro-osteogenic factor related with AV calcification [21], and Runx2 is an essential transcription factor associated with osteoblast differentiation [38]. In this study, expression of BMP2 and Runx2 was up-regulated after stimulation with AGE-BSA, increasing synthesis of bone matrix protein, osteopontin. Introduction of siRNA to knock down RAGE expression blocked this effect. This implied that RAGE activation is associated with osteoblastic differentiation of VICs.
An abundance of studies have confirmed the crucial role of chronic inflammation in the pathogenesis of AV calcification. Humans and animals with AV calcification both show markedly elevated inflammatory cells infiltration and production of pro-inflammatory cytokines. Normal AV are devoid of inflammatory cells, but, calcified valves have large numbers of accumulated cells including macrophages, T lymphocytes and mast cells [14, 36, 37, 45]. Inflammation-associated molecules, IL-1β, 5-lipoxygenase, MCP-1, TNF-α have also been identified in calcified valves and they have been correlated with the severity of AV calcification [22, 23, 32].
Observations and experimental evidence strongly suggest that RAGE signaling results in profound inflammation [9, 26]. Previous studies have indicated that the vascular and inflammatory stresses mediated by interaction between RAGE and its ligands, such as AGEs, play a central role in the development of accelerated atherosclerosis [13]. Strategies targeting to inactivate RAGE promote plaque stabilization and inhibit the progression of atherosclerotic changes [29, 41, 44]. Our data support the conclusion that, RAGE-mediated pro-inflammatory pathway is involved not only in atherosclerosis but also in AV calcification. We have shown that, RAGE activation is accompanied by enhanced inflammatory cells infiltration in the calcified AV in hypercholesterolemic rabbits.
Our in vitro data demonstrated that AGE-BSA activated RAGE and induced RAGE-mediated signaling pathways in VICs, subsequently evoking the increased production of pro-inflammatory cytokines IL-8, TNF-α and MCP-1. We also observed that RAGE siRNA suppressed AGE-BSA-stimulated up-regulation of these cytokines. In summary, RAGE activation in VICs induced inflammatory consequences. The role of TNF-α in inflammation-induced calcification has been confirmed, as TNF-α neutralizing antibody, infliximab, inhibits aortic BMP2-Msx-Wnt calcification cascade in Ldlr−/− mice [1]. In addition, TNF-α accelerates the calcification of VICs from patients with AV calcification [51]. MCP-1 has also been associated with macrophages infiltration in a rabbit model of AV calcification model [42]. These data suggest that inflammation appears to be a key mediator between RAGE activation and AV calcification.
Activation of RAGE relays cell surface signals to various intracellular pathways including the NF-κB pathway [47]. NF-κB pathway is a canonical pathway responsible for the production of pro-inflammatory cytokines. The key step in NF-κB activation is the stimulus-dependent, residue-specific phosphorylation of NF-κB inhibitors, IκBs. In our experiment, we showed that RAGE activation induced IκBα degradation and enhanced NF-κB p65 intranuclear translocation. Pre-treatment with NF-κB inhibitors Bay 11-7082 significantly inhibited RAGE-mediated up-regulation of Runx2 and osteopontin. These results indicate that NF-κB activation mediates AGE-BSA-induced pro-inflammatory effects and osteoblastic differentiation of VICs. Pioglitazone also inhibited NF-κB activation. However, there are data also supporting a crosstalk between the canonical NF-κB and Notch signaling pathways inhibits PPARγ expression and promotes pancreatic cancer progression [30].
Our in vivo data demonstrated several beneficial effects of pioglitazone administration on the AV, including attenuated progression of AV calcification, reduced inflammatory cells infiltration, and reduced oxidative stress. Previous studies have already shown the protective effect against some cardiovascular pathology [4, 18, 50]. Joner et al. [20] demonstrated that oral administration of pioglitazone suppressed in-stent neointimal growth by limiting local inflammatory pathways. Similarly, we found that pioglitazone inhibited AGE-BSA-induced inflammation and osteoblastic differentiation within VICs in vitro. All these effects were concomitantly accompanied with reduction of RAGE expression. As demonstrated in the present study, suppression of RAGE activation is probably a molecular target of attenuated AV calcification after pioglitazone treatment. Previous studies do support the conclusion that PPARγ agonists lower the expression of RAGE both in vivo and in vitro [18, 31, 49]. Wang et al. [49] reported that PPARγ agonists suppressed neointimal hyperplasia after balloon angioplasty in both diabetic and non-diabetic rats, and reduced RAGE expression and S100-induced SMC proliferation. Marx et al. [31] reported that rosiglitazone and pioglitazone diminished RAGE protein expression in human endothelial cells, decreasing AGE-BSA and β-amyloid-induced MCP-1 production. The present study extends previous findings. To our knowledge, this is the first to show favorable effects of pioglitazone against AV calcification. These effects are contributed to pioglitazone’s ability to inhibit RAGE activation and inflammatory responses. The mechanism underlying the down-regulation of RAGE expression by pioglitazone was not investigated in this study.
In conclusion, the present study highlights the detrimental role of RAGE activation and RAGE signaling in the development and progression of AV calcification. Our data suggest that RAGE activation in VICs results in enhanced production of pro-inflammatory cytokines and osteoblast-like differentiation. Oral pioglitazone treatment in hypercholersterolemic rabbits caused significant alleviation of AV calcification. This beneficial effect may be explained in part by targeted reduction of RAGE activation and reduced inflammatory response. Our data provide new insight into how PPARγ agonists, might slow the development of AV calcification. Further studies are warranted to elucidate other possible mechanisms.
Limitations
For both ethical reasons and the cell growth rate, we used porcine instead of human aortic VICs. Some of these results may not be able to explain the actual mechanisms underlying this or any process in human. We showed that pioglitazone inhibited AGE-BSA-induced NF-κB activation in VICs, but one previous study did not show pioglitazone to have any effect on NF-κB in humans [12]. This indicates that this effect may only take place in other animals or in vitro.
References
Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA (2007) Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol 27:2589–2596. doi:10.1161/ATVBAHA.107.153668
Basta G, Corciu AI, Vianello A, Del Turco S, Foffa I, Navarra T, Chiappino D, Berti S, Mazzone A (2010) Circulating soluble receptor for advanced glycation end-product levels are decreased in patients with calcific aortic valve stenosis. Atherosclerosis 210:614–618. doi:10.1016/j.atherosclerosis.2009.12.029
Beckmann E, Grau JB, Sainger R, Poggio P, Ferrari G (2010) Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis 19:441–452
Birnbaum Y, Long B, Qian J, Perez-Polo JR, Ye Y (2011) Pioglitazone limits myocardial infarct size, activates Akt, and upregulates cPLA2 and COX-2 in a PPAR-gamma-independent manner. Basic Res Cardiol 106:431–446. doi:10.1007/s00395-011-0162-3
Bonow RO, Carabello BA, Chatterjee K, De Leon AC Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O’Gara PT, O’Rourke RA, Otto CM, Shah PM, Shanewise JS (2008) 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American college of Cardiology/American heart association task force on practice guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease): endorsed by the society of cardiovascular anesthesiologists, society for cardiovascular angiography and interventions, and society of thoracic surgeons. Circulation 118:e523–e661. doi:10.1161/CIRCULATIONAHA.108.190748
Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, Migheli A, Stern D (1993) Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143:1699–1712
Cai Z, Li F, Peng C, Li H, Zong Y, Liu Z, Qu S (2010) Effect of insulin on the differential expression of VLDL receptor isoforms of SGC7901 cell and its biological implication. J Huazhong Univ Sci Technol Med Sci 30:551–555. doi:10.1007/s11596-010-0541-2
Cecil DL, Terkeltaub RA (2011) Arterial calcification is driven by RAGE in Enpp 1−/− mice. J Vasc Res 48:227–235. doi:10.1159/000318805
Chavakis T, Bierhaus A, Nawroth PP (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 6:1219–1225. doi:10.1016/j.micinf.2004.08.004
Drolet MC, Arsenault M, Couet J (2003) Experimental aortic valve stenosis in rabbits. J Am Coll Cardiol 41:1211–1217
Gallagher AM, Smeeth L, Seabroke S, Leufkens HG, van Staa TP (2011) Risk of death and cardiovascular outcomes with thiazolidinediones: a study with the general practice research database and secondary care data. PLoS ONE 6:e28157. doi:10.1371/journal.pone.0028157
Hanefeld M, Pfutzner A, Forst T, Kleine I, Fuchs W (2011) Double-blind, randomized, multicentre, and active comparator controlled investigation of the effect of pioglitazone, metformin, and the combination of both on cardiovascular risk in patients with type 2 diabetes receiving stable basal insulin therapy: the PIOCOMB study. Cardiovasc Diabetol 10:65. doi:10.1186/1475-2840-10-65
Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM (2008) Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest 118:183–194. doi:10.1172/JCI32703
Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT (2004) Induction of local angiotensin II-producing systems in stenotic aortic valves. J Am Coll Cardiol 44:1859–1866. doi:10.1016/j.jacc.2004.07.054
Hernandez AV, Usmani A, Rajamanickam A, Moheet A (2011) Thiazolidinediones and risk of heart failure in patients with or at high risk of type 2 diabetes mellitus: a meta-analysis and meta-regression analysis of placebo-controlled randomized clinical trials. Am J Cardiovasc Drugs 11:115–128. doi:10.2165/11587580-000000000-00000
Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, Earley J, McNally EM (2011) S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol 31:337–344. doi:10.1161/ATVBAHA.110.217745
Hsueh WA, Bruemmer D (2004) Peroxisome proliferator-activated receptor gamma: implications for cardiovascular disease. Hypertension 43:297–305. doi:10.1161/01.HYP.0000113626.76571.5b
Ihm SH, Chang K, Kim HY, Baek SH, Youn HJ, Seung KB, Kim JH (2010) Peroxisome proliferator-activated receptor-gamma activation attenuates cardiac fibrosis in type 2 diabetic rats: the effect of rosiglitazone on myocardial expression of receptor for advanced glycation end products and of connective tissue growth factor. Basic Res Cardiol 105:399–407. doi:10.1007/s00395-009-0071-x
Johnson CM, Hanson MN, Helgeson SC (1987) Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J Mol Cell Cardiol 19:1185–1193
Joner M, Farb A, Cheng Q, Finn AV, Acampado E, Burke AP, Skorija K, Creighton W, Kolodgie FD, Gold HK, Virmani R (2007) Pioglitazone inhibits in-stent restenosis in atherosclerotic rabbits by targeting transforming growth factor-beta and MCP-1. Arterioscler Thromb Vasc Biol 27:182–189. doi:10.1161/01.ATV.0000251021.28725.e8
Kaden JJ, Bickelhaupt S, Grobholz R, Vahl CF, Hagl S, Brueckmann M, Haase KK, Dempfle CE, Borggrefe M (2004) Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J Heart Valve Dis 13:560–566
Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, Kilic R, Sarikoc A, Pinol R, Hagl S, Lang S, Brueckmann M, Borggrefe M (2005) Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol 14:80–87. doi:10.1016/j.carpath.2005.01.002
Kaden JJ, Dempfle CE, Grobholz R, Tran HT, Kilic R, Sarikoc A, Brueckmann M, Vahl C, Hagl S, Haase KK, Borggrefe M (2003) Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis 170:205–211
Katz R, Budoff MJ, Takasu J, Shavelle DM, Bertoni A, Blumenthal RS, Ouyang P, Wong ND, O’Brien KD (2009) Relationship of metabolic syndrome with incident aortic valve calcium and aortic valve calcium progression: the Multi-Ethnic Study of Atherosclerosis (MESA). Diabetes 58:813–819. doi:10.2337/db08-1515
Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J Jr, Pomerantzeff PM, Laurindo FR (2008) Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol 28:463–470. doi:10.1161/ATVBAHA.107.156745
Lin L (2006) RAGE on the Toll Road? Cell Mol Immunol 3:351–358
Lin L, Park S, Lakatta EG (2009) RAGE signaling in inflammation and arterial aging. Front Biosci 14:1403–1413
Loke YK, Kwok CS, Singh S (2011) Comparative cardiovascular effects of thiazolidinediones: systematic review and meta-analysis of observational studies. BMJ 342:d1309. doi:10.1136/bmj.d1309
Lu Y, Qin W, Shen T, Dou L, Man Y, Wang S, Xiao C, Li J (2011) The antioxidant N-acetylcysteine promotes atherosclerotic plaque stabilization through suppression of RAGE, MMPs and NF-kappaB in ApoE-deficient mice. J Atheroscler Thromb 18:998–1008
Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, Balkwill FR, Tuveson DA, Hagemann T (2011) Crosstalk between the canonical NF-kappaB and Notch signaling pathways inhibits Ppargamma expression and promotes pancreatic cancer progression in mice. J Clin Invest 121:4685–4699. doi:10.1172/JCI45797
Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, Hombach V, de Caterina R, Basta G, Wautier MP, Wautiers JL (2004) Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 53:2662–2668
Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, Hansson GK, Back M (2011) Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation 123:1316–1325. doi:10.1161/CIRCULATIONAHA.110.966846
Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004
Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M (2006) Burden of valvular heart diseases: a population-based study. Lancet 368:1005–1011. doi:10.1016/S0140-6736(06)69208-8
O’Brien KD (2006) Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more). Arterioscler Thromb Vasc Biol 26:1721–1728. doi:10.1161/01.ATV.0000227513.13697.ac
Olsson M, Dalsgaard CJ, Haegerstrand A, Rosenqvist M, Ryden L, Nilsson J (1994) Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. J Am Coll Cardiol 23:1162–1170
Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD (1994) Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 90:844–853
Prince M, Banerjee C, Javed A, Green J, Lian JB, Stein GS, Bodine PV, Komm BS (2001) Expression and regulation of Runx2/Cbfa1 and osteoblast phenotypic markers during the growth and differentiation of human osteoblasts. J Cell Biochem 80:424–440. doi:10.1002/1097-4644(20010301)80:3<424
Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM (2011) Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease-2011 update. Circulation 124:1783–1791. doi:10.1161/CIRCULATIONAHA.110.006767
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391:79–82. doi:10.1038/34178
Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, Barit D, Coughlan MT, Drew BG, Lancaster GI, Thomas M, Forbes JM, Nawroth PP, Bierhaus A, Cooper ME, Jandeleit-Dahm KA (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469. doi:10.2337/db07-1808
Speidl WS, Cimmino G, Ibanez B, Elmariah S, Hutter R, Garcia MJ, Fuster V, Goldman ME, Badimon JJ (2010) Recombinant apolipoprotein A-I Milano rapidly reverses aortic valve stenosis and decreases leaflet inflammation in an experimental rabbit model. Eur Heart J 31:2049–2057. doi:10.1093/eurheartj/ehq064
Suga T, Iso T, Shimizu T, Tanaka T, Yamagishi S, Takeuchi M, Imaizumi T, Kurabayashi M (2011) Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb 18:670–683
Sun L, Ishida T, Yasuda T, Kojima Y, Honjo T, Yamamoto Y, Yamamoto H, Ishibashi S, Hirata K, Hayashi Y (2009) RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc Res 82:371–381. doi:10.1093/cvr/cvp036
Syvaranta S, Helske S, Laine M, Lappalainen J, Kupari M, Mayranpaa MI, Lindstedt KA, Kovanen PT (2010) Vascular endothelial growth factor-secreting mast cells and myofibroblasts: a novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arterioscler Thromb Vasc Biol 30:1220–1227. doi:10.1161/ATVBAHA.109.198267
Tanikawa T, Okada Y, Tanikawa R, Tanaka Y (2009) Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vasc Res 46:572–580. doi:10.1159/000226225
Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, Andrassy J, Bierhaus A, Kaya Z, Katus HA, Andrassy M (2012) S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol 107:250. doi:10.1007/s00395-012-0250-z
Volz HC, Seidel C, Laohachewin D, Kaya Z, Muller OJ, Pleger ST, Lasitschka F, Bianchi ME, Remppis A, Bierhaus A, Katus HA, Andrassy M (2010) HMGB1: the missing link between diabetes mellitus and heart failure. Basic Res Cardiol 105:805–820. doi:10.1007/s00395-010-0114-3
Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, Qu W, Lincoff AM, Schmidt AM, Topol EJ, Penn MS (2006) Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther 317:37–43. doi:10.1124/jpet.105.095125
Ye Y, Perez-Polo JR, Aguilar D, Birnbaum Y (2011) The potential effects of anti-diabetic medications on myocardial ischemia–reperfusion injury. Basic Res Cardiol 106:925–952. doi:10.1007/s00395-011-0216-6
Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa K (2011) Tumor necrosis factor-alpha accelerates the calcification of human aortic valve interstitial cells obtained from patients with calcific aortic valve stenosis via the BMP2-Dlx5 pathway. J Pharmacol Exp Ther 337:16–23. doi:10.1124/jpet.110.177915
Acknowledgments
This work was funded by grants from the National Natural Science Foundation of China (81170214) and “863” Program (2009AA03Z420). We specially appreciate the great help with echocardiography from Dr. Xiaojun Bi.
Author information
Authors and Affiliations
Corresponding authors
Additional information
F. Li and Z. Cai contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Li, F., Cai, Z., Chen, F. et al. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation end products. Basic Res Cardiol 107, 306 (2012). https://doi.org/10.1007/s00395-012-0306-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0306-0