
Abstract
Introduction
Although meningiomas are frequently diagnosed in adults, it is a rare (intracranial) tumor in the pediatric population, with an incidence of 0.06/100,000. The pathology and treatment of meningiomas in adulthood has been a topic of increasing investigation. So far, the treatment of pediatric meningiomas has been extrapolated from these results. The question remains, however, whether translation of adult meningioma data into the childhood population is legitimate.
Methods
We present the case of a 3-year-old girl diagnosed with an intraventricular malignant meningioma and type 2 neurofibromatosis. She was operated on multiple times to achieve complete resection and received adjuvant chemotherapy. Since, she has been stable with no neurological sequelae and/or recurrence of the meningioma.
Conclusion
Pediatric meningiomas are rare tumors and differ from their adult counterparts in various aspects. We believe that gross total resection of meningioma in the pediatric population, when possible, is the treatment of choice. In the event of a subtotal resection, repeat resection is recommended. Any adjuvant treatment with chemotherapy or radiation therapy should be carefully considered during multidisciplinary meetings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Meningiomas in childhood are rare intracranial tumors as opposed to meningiomas occurring in adulthood (24–30 % of CNS tumors) [12]. In a large meta-analysis covering published meningioma data from the year 1948 until 2007, Kotecha et al. could only identify 677 children and adolescents with meningioma [13]. Childhood meningioma constituted 2.2 % of pediatric CNS tumors [13]. In a recent Dutch study, the calculated incidence of operated childhood meningiomas was as low as 0.06/100,000 children [23]. To date, the genetic and molecular background of meningiomas is studied extensively and results have posed more targeted therapies [1]. These results, however, are most often obtained from meningiomas in adult patients and extrapolated to the pediatric population [24]. It is debated whether this approach is legitimate, since remarkable differences in childhood and adult meningioma have been observed besides its lower incidence. Interestingly, for example, childhood meningioma shows a more malignant course, as illustrated by pathology and overall survival [12, 13, 23, 24]. For this reason, we present the case of a 3-year-old girl diagnosed with two intraventricular meningiomas and review the literature and treatment of meningiomas in the pediatric population.
Case presentation
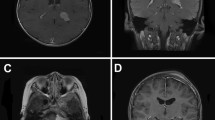
A Caucasian 3-year-old girl was referred to our clinic with gait disturbances and progressive lethargy since 4 weeks in June 2010. She had no complaints of headache, nausea, or vomiting and her antecedents were unremarkable. Physical examination revealed papilledema and no clinical signs of neurofibromatosis. The referring neurologist performed a computed tomography scan that showed a large intraventricular tumor with accompanying hydrocephalus. An MRI scan showed a large intraventricular tumor 72 × 48 × 43 mm in size, located in the right lateral ventricle expanding into the foramen of Monro and a smaller separate lesion in the right temporal horn, subsequently causing an obstructive hydrocephalus with transependymal effusion suggestive of increased intracranial pressure (Fig. 1). The differential diagnosis included a choroid plexus carcinoma/papilloma, meningioma, or ependymoma.

Preoperative MRI. Sagittal non-contrast-enhanced T1 MRI (a) and axial contrast-enhanced T1 MRI (b, c)
Four days after admission, she was operated through a transcortical approach via the right superior frontal gyrus. A macroscopically complete resection of the lesion in the right lateral ventricle was thought to be achieved. An MRI within 48 h after surgery showed postoperative effects and, to our surprise, a residual lesion (20 × 18 × 17 mm) in the third ventricle as well as the untouched lesion in the right temporal horn (Fig. 2a).

a Postoperative MRI scan 2 days after initial surgery, contrast-enhanced axial T1 showing a residual lesion in the third ventricle as well as a lesion in the right temporal horn. b Contrast-enhanced axial T1 MRI 3 months after starting hydroxyurea therapy, which showed a tenfold increase in tumor volume. c Contrast-enhanced axial T1 MRI after the second surgery on tumor in the lateral ventricle and showing a stable lesion in the right temporal horn. d Contrast-enhanced sagittal T1 MRI of vertebral column showing a lesion at the level of C1. e, f Follow-up MRI scan in November 2013 showing no residual lesions intraventricularly
She developed a mild transient left-sided hemiparesis that recovered after 1 day. The pathology report showed an atypical meningioma, WHO grade II (Fig. 3, left panel), for which a wait-and-scan policy was followed, as discussed in our local pediatric neuro-oncology board. The patient was considered too young for radiotherapy treatment at that time.

Left panel HE × 250 depicts an atypical meningioma, WHO II. Right panel HE × 250 depicts a malignant meningioma, WHO III
Subsequently, an MRI scan of the cerebrum and neuraxis was performed 4 weeks later. This MRI showed an increase in the lesion near the foramen of Monro, no significant change in size of the lesion in the right temporal horn, and some evidence of leptomeningeal spread at the C1 level. Considering a poor prognosis, we decided to start systemic chemotherapy treatment with hydroxyurea up to a dose of 20 mg kg−1. At this point, also genetic analysis for neurofibromatosis type II was performed.
After 3 months, the treatment was evaluated with MRI which, unfortunately, again showed a tenfold increase in the volume of the right lateral intraventricular tumor from 22 × 21 × 28 to 53 × 49 × 50 mm (Fig. 2b).
The tumor in the right temporal horn and the suspicious lesion at the C1 level remained unchanged. Despite all this, the patient remained neurological unremarkable. She was therefore again operated on via the same approach in a two-stage surgery. The postoperative MRI in the following day showed no signs of residual tumor in the right lateral or third ventricle. The postoperative course was complicated by diabetes insipidus and epilepsy related to an increased intracranial pressure. This resolved after placement of an external ventricular shunt. The diabetes insipidus was treated with Minrin and sodium supplements, and no antiseizure medication was indicated. No permanent cerebrospinal fluid shunting was needed. The pathology report now revealed a malignant meningioma, WHO grade III (Fig. 3, right panel). After recovery, the hydroxyurea therapy was ceased and she was treated with etoposide and carboplatin for 21 months.
Four weeks after surgery, an MRI scan showed the same aspect and size of the tumor in the right temporal horn, and no residual or regrowth in the right lateral ventricle (Fig. 2c). Because of this, we decided to operate on the tumor in the right temporal horn via a transcortical approach through the inferior temporal gyrus 3 months after the last surgery. The pathology report showed a WHO grade II meningioma. The postoperative MRI showed no residual tumor, and the lesion at the C1 level, suspicious for leptomeningeal metastasis, had again not changed (Fig. 2d).
The chemotherapy treatment was scheduled to continue until the age of 5 years, at which age she would be amenable for radiotherapy on the C1 lesion, which had remained stable. She was, however, diagnosed with neurofibromatosis type 2 (NF2) in August 2012. Because of this diagnosis, we decided not to go through with the radiotherapy and stop the chemotherapy treatment. A further wait-and-scan approach had been followed since. At the last follow-up in November 2013, the MRI showed no regrowth of the intraventricular meningiomas and she remains without neurological symptoms (Fig. 2e, f).
Discussion
We present the case of a 3-year-old girl diagnosed with an intraventricular malignant meningioma, WHO grade III, and type 2 neurofibromatosis. She was operated on multiple times to achieve complete resection and received adjuvant chemotherapy. Since, she has been stable with no neurological sequelae and/or recurrence of the meningioma.
Meningiomas constitute 0.4–4.6 % of pediatric central nervous tumors, whereas meningiomas account for 30 % of all primary central nervous tumors in adults [12]. Childhood meningiomas account for 1.5–2 % of all intracranial meningiomas [10, 25]. In adults, they are twice as common in women as in men (1.7:1), whereas in children, there is a slight male predominance (1.3:1) [25, 2]. Known risk factors for pediatric meningiomas are neurofibromatosis type 2 and previous radiation therapy [1, 12, 13, 23, 24]. Twenty to 40 % of pediatric meningiomas develop in patients with neurofibromatosis type 2 [24]. Also, Gorlin syndrome (multiple basal cell carcinoma syndrome), an autosomal dominant inherited familial cancer syndrome, is associated with pediatric meningioma [20, 24]. Furthermore, meningiomas can be associated with Rubinstein-Taybi syndrome [18]. Radiation therapy is a well-known causative factor of meningiomas but is less common in children than in adults. The time interval between exposure to radiation therapy and developing meningioma is less than 20 years after high-dose radiation (>20 Gy) and up to 35 years after low-dose radiation (8 Gy) [24].
Molecular mechanisms are poorly defined in the pediatric population. The initial cytogenetic event leading to a meningioma is thought to be loss of genetic material of the long arm of chromosome 22, which is close to the neurofibromatosis type 2 gene (22q12) [7]. Furthermore, 1p and 14q deletions are frequently found in pediatric meningiomas and are associated with higher recurrence risk [24, 27].
Arivazhagan et al. [2] compared pediatric meningiomas to their adult counterparts. They performed a retrospective analysis of 33 patients under the age of 18 years over a 16-year period. A feature that distinguished the pediatric from the adult population was tumor location. Whereas convexity meningiomas are commonest in the adult population, intraventricular and skull base tumors are more common in children. Liu et al. [15] reported in a series of 675 adult meningiomas only 3.7 % intraventricular localizations, compared to 19.4 % in four pediatric series of 98 combined cases [2, 15–17, 19]. Burkhardt et al. [3] described 12 patients under the age of 18 years treated for meningioma, comparing skull base meningioma to non-skull base meningioma. They concluded that skull base meningiomas were smaller in size but were less likely to be completely resected [3]. Furthermore, there is a higher rate of malignancy in pediatric meningiomas and cystic meningiomas occur more often in children than in adults [8, 25]. These results were corroborated by Thuijs et al. [23].
Santos et al. [21] stated that location of tumor, associated factors such as radiotherapy and neurofibromatosis type 2, and extent of excision appear to be more important than histology in predicting outcome.
Published data regarding the treatment of meningiomas in the pediatric population are few, and there are no validated guidelines for managing pediatric meningiomas. Most of the data are extrapolated from studies done in the adult population. The question however remains whether extrapolating results from the adult population to a pediatric population is correct and legitimate.
Total resection of meningiomas in children has been challenging because of several factors such as location, size, and smaller blood volume in children. Kotecha et al. [13] performed a meta-analysis of meningiomas in children and adolescents. Their analysis showed that from a total of 677 children, those who underwent initial gross total resection had a better relapse free survival (RFS) and overall survival (OS) than those who had a subtotal resection (RFS: gross total resection vs subtotal resection, 5 years, 85.8 vs 46 %; 10 years, 79.7 vs 32.4 %; and 15 years, 78.4 vs 10.8 %; OS: gross total resection vs subtotal resection, 5 years, 95 vs 77.3 %; 10 years, 89.8 vs 54.3 %; and 15 years, 81.1 vs 48.3 %) [13]. Furthermore, children with NF2 had a significantly higher risk of multifocal meningioma and had significantly worse RFS and OS (RFS: none vs NF2, 5 years, 77.3 vs 72.4 %; 10 years, 71.8 vs 49.4 %; and 15 years, 67.8 vs 49.4 %; OS: none vs NF2, 5 years, 89.7 vs 97.9 %; 10 years, 81.9 vs 80.0 %; and 15 years, 77.4 vs 57.9 %) [13].
The use of adjuvant radiotherapy in children below 3–5 years remains controversial, due to the risk of late sequelae [1]. Kotecha et al. and Traunecker et al. [13, 24] found no successful chemotherapeutical options. Chamberlain [4] reviewed the current chemotherapy for recurrent intracranial meningiomas in adult patients. At present, expert guidelines recommend three agents: hydroxyurea, interferon alpha, and long-acting somatostatin. Hydroxyurea is an oral ribonucleotide reductase inhibitor and arrests meningioma cell growth through arrest of the S phase of the cell cycle, thus inducing apoptosis [22]. Chamberlain reported stable disease in 35 % and progressive disease in 65 % of patients with hydroxyurea [4]. Kim et al. [11] reported the long-term follow-up result of hydroxyurea therapy in patients with recurrent meningiomas. They concluded that hydroxyurea is well tolerated and can induce long-term stabilization of disease, but that it has modest effect against recurrent meningiomas [11].
Interferon alpha inhibits the growth of cultured human meningioma cell lines in vitro. Four small studies have been published of which the largest study had 35 patients with recurrent, unresectable, and previously radiated low-grade meningioma (WHO grade I) who were treated with interferon alpha [5]. In this group, no radiographic response was seen; however, 74 % showed stable disease with a median progression-free survival (PFS) of 7 months. Median overall survival was 8 months, suggesting that interferon alpha is effective for recurrent low-grade meningioma [5].
Many tumors express somatostatin receptors. Among brain tumors, meningiomas have shown the highest frequency in somatostatin receptor expression detected by octreotide scintigraphy (up to a 90 % positive rate of detection) [6]. The largest trial of somatostatin use in meningiomas performed in 16 patients with recurrent meningiomas by Chamberlain et al. showed that 31 % of patients had partial radiographic response and 44 % achieved PFS at 6 months with minimal toxicity [6].
Our patient was first treated with hydroxyurea and later with etoposide and carboplatin. She never received radiotherapy because she was too young and was later diagnosed with NF2.
Etoposide or VP16 is derived from a plant alkaloid (podophyllotoxin) and is thought to block an enzyme topoisomerase II, which leads to breaks in DNA strands which leads to cell death. Carboplatin is an alkylating agent, which damages the DNA inside cancerous cells and causes cell death. The combination chemotherapy of etoposide and carboplatin has mostly been used in the adult population as treatment for different types of cancer (lung cancer, testicular cancer, ovarian cancer) and as treatment for different types of brain tumors as in recurrent malignant gliomas [26]. In the pediatric population, it has shown some beneficial effect in small groups with choroid plexus carcinomas or PNET [9, 14]. However, there has not been any publication of this combination with recurrent meningiomas. Despite the lack of experience with this combination of chemotherapy in recurrent meningiomas, our patient was treated with etoposide/carboplatin because of good experience in other difficult-to-treat tumors. It showed an enormous beneficial effect in our patient in stabilizing the disease after achieving gross total resection.
In conclusion, pediatric meningiomas are rare tumors and differ from their adult counterparts in various aspects. Gross total resection of meningioma in the pediatric population remains to be the ultimate therapeutic goal. In the event of a subtotal resection, repeat resection is recommended. Any adjuvant treatment with chemotherapy or radiation therapy of pediatric recurrent meningioma should be carefully considered during multidisciplinary meetings.
References
Alexiou GA, Markoula S, Gogou P, Kyritsis AP (2011) Genetic and molecular alterations in meningiomas. Clin Neurol Neurosurg 113:261–267. doi:10.1016/j.clineuro.2010.12.007
Arivazhagan A, Devi BI, Kolluri SVR, Abraham RG, Sampath S, Chandramouli BA (2008) Pediatric intracranial meningiomas—do they differ from their counterparts in adults? Pediatr Neurosurg 44:43–48
Burkhardt JK, Neidert MC, Grotzer MA, Krayenbűhl N, Bozinov O (2013) Surgical resection of pediatric skull base meningiomas. Child Nerv Syst 29:83–87. doi:10.1007/s00381-012-1906-6
Chamberlain MC (2012) The role of chemotherapy and targeted therapy in the treatment of intracranial meningioma. Curr Opin Oncol 24:666–671. doi:10.1097/CCO.0b013e328356364d
Chamberlain MC, Glantz MJ (2008) α-Interferon for recurrent WHO grade I intracranial meningiomas. Cancer 113:2146–2151. doi:10.1002/cncr.23803
Chamberlain MC, Glantz MJ, Fadul CE (2007) Recurrent meningioma: salvage therapy with Sandostatin. Neurology 69:969–973
Cogen PH, Daneshvar L, Bowcock AM (1991) Loss of heterozygosity for chromosome 22 DNA sequences in human meningioma. Cancer Genet Cytogenet 53:271–277
Ferrante L, Acqui M, Artico M, Mastronardi L, Rocchi G, Fortuna A (1989) Cerebral meningiomas in children. Childs Nerv Syst 5:83–86
Friedrich C, von Bueren AO, von Hoff K, Gerber NU, Ottensmeier H, Deinlein F et al (2013) Treatment of young children with CNS-primitive neuroectodermal tumors/pineoblastomas in the prospective multicenter trial HIT 2000 using different chemotherapy regimens and radiotherapy. Neuro Oncol 15(2):224–234. doi:10.1093/neuonc/nos292
Gao X, Zhang R, Mao Y, Wang Y (2009) Childhood and juvenile meningiomas. Childs Nerv Syst 25(12):1571–1580. doi:10.1007/s00381-009-0964-x
Kim MS, Yu DW, Jung YJ, Kim SW, Chang CH, Kim OL (2012) Long-term follow-up result of hydroxyurea chemotherapy for recurrent meningiomas. J Korean Neurosurg Soc 52(6):517–522. doi:10.3340/jkns.2012.52.6.517
Kotecha RS, Junckerstorff RC, Lee S, Cole CH, Gottardo NG (2011) Pediatric meningioma: current approaches and future direction. J Neurooncol 104:1–10. doi:10.1007/s11060-010-0503-3
Kotecha RS, Pascoe EM, Rushing EJ, Rorke-Adams LB, Zwerdling T, Gao X et al (2011) Meningiomas in children and adolescents: a meta-analysis of individual patient data. Lancet Oncol 12:1229–1239. doi:10.1016/S1470-2045(11)70275-3
Lafay-Cousin L, Mabbott DJ, Halliday W, Taylor MD, Tabori U, Kamaly-Asl ID et al (2010) Use of ifosfamide, carboplatin and etoposide chemotherapy in choroid plexus carcinoma. A clinical article. J Neurosurg Pediatrics 5:615–621. doi:10.3171/2010.3.PEDS09354
Liu M, Wei Y, Liu Y, Zhu S, Li X (2006) Intraventricular meningiomas: a report of 25 cases. Neurosurg Rev 29:36–40
Malucci CL, Parkes SE, Barber P, Powell J, Stevens MC, Walsh AR et al (1996) Paediatric meningeal tumours. Childs Nerv Syst 12:582–589
Mehta N, Bhagwati S, Parulekar G (2009) Meningiomas in children: a study of 18 cases. J Pediatr Neurosci 4:61–65. doi:10.4103/1817-1745.57322
Miller RW, Rubinstein LJ (1995) Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 56:112–115
Okechi H, Albright AL (2012) Intraventricular meningioma: case report and literature review. Pediatr Neurosurg 48:30–34. doi:10.1159/000341176
Rushing EJ, Olsen C, Mena H, Rueda ME, Lee YS, Keating RF et al (2005) Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg 103:489–495
Santos MV, Furlanetti L, Valera ET, Brassesco MS, Tone LG, Santos de Oliveira R (2012) Pediatric meningiomas: a single-center experience with 15 consecutive cases and review of the literature. Childs Nerv Syst 28:1887–1896. doi:10.1007/s00381-012-1823-8
Sherman WJ, Raizer JJ (2012) Chemotherapy: what is its role in meningioma? Expert Rev Neurother 12(10):1189–1196. doi:10.1586/ern.12.108
Thuijs NB, Uitdehaag BMJ, van Ouwerkerk WJR, van der Valk P, Vandertop WP, Peerdeman SM (2012) Pediatric meningiomas in the Netherlands 1974-2010: a descriptive epidemiological case study. Childs Nerv Syst 28:1009–1015. doi:10.1007/s00381-012-1759-z
Traunecker H, Malucci C, Grundy R, Pizer B, Saran F (2008) Children’s Cancer and Leukaemia Group (CCLG): guidelines for the management of intracranial meningioma in children and young people. Br J Neurosurg 22(1):13–25. doi:10.1080/02688690701842208
Tufan K, Dogulu F, Kurt G, Emmez H, Ceviker N, Baykaner MK (2005) Intracranial meningiomas of childhood and adolescence. Pediatr Neurosurg 41:1–7
Watanabe K, Kanaya H, Fujiyama Y, Kim P (2002) Combination chemotherapy using Carboplatin (JM-8) and etoposide (JET therapy) for recurrent malignant gliomas: a phase II study. Acta Neurochir 144:1265–1270
Weber RG, Boström J, Wolter M, Baudis M, Collins VP, Reifenberger G et al (1997) Analysis of genomic alterations in benign, atypical and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci 94:14719–14724
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kwee, L.E., van Veelen-Vincent, M.L.C., Michiels, E.M.C. et al. The importance of microsurgery in childhood meningioma: a case report. Childs Nerv Syst 31, 161–165 (2015). https://doi.org/10.1007/s00381-014-2490-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-014-2490-8