
Abstract
With improvement in leukemia therapy, central nervous system (CNS) tumors are the leading cause of cancer mortality in children and the most expensive of all human neoplasms to treat. Meningiomas are rare intracranial tumors in childhood and adolescence arising from arachnoid cell clonal outgrowth in the meninges. There have been no collaborative prospective therapeutic trials for pediatric meningioma because of its rarity, and the best evidence for management comes from retrospective case analyses and extrapolation from the treatment of adult meningioma. However this may not be ideal, because the underlying biology of adult and pediatric meningiomas seems to be different, as is the case for other CNS tumors. In addition, treatment of pediatric brain tumors requires consideration of long-term quality of life. This review reflects on what is currently known about pediatric meningiomas and opportunities for future directions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Meningiomas constitute 0.4–4.6% of pediatric central nervous system (CNS) tumors [1]. In marked contrast, meningiomas account for approximately 30% of all primary CNS tumors in adults. Only 1.5–2.0% of all meningiomas occur in children [1]. They are twice as common in women as in men [2], whereas in children there is a slight male predominance. The incidence of pediatric meningioma increases with age [3] and the mean age at diagnosis is greater than that of other pediatric CNS tumors [4]. Infantile meningiomas are exceedingly rare [5]. The 2010 Central Brain Tumor Registry of the United States statistical report for tumors diagnosed between 2004 and 2006, recorded a total of 285 pediatric meningiomas, with 84 cases occurring in the first decade of life and 201 in the second decade, whereas in adults 53,170 meningiomas were recorded [6].
Etiology
A number of factors are known to contribute to pediatric meningioma. Neurofibromatosis type 2 (NF2) and ionizing radiation are well known risk factors and are discussed in detail below. Rare associations include Gorlin syndrome, especially in those who have previously received cranial radiotherapy [7–9]. Gorlin syndrome, also known as multiple basal cell carcinoma syndrome, is an autosomal dominant condition, due to mutations in the patched (PTCH) gene on chromosome 9, and involves defects within multiple body systems. There is one report of seven children with chordoid meningiomas and systemic manifestations of Castleman’s disease [10], a rare, lymphoproliferative disorder characterized by non-malignant tumors at a single or multiple sites throughout the body. Meningiomas have been associated with Rubenstein–Taybi syndrome [11]. Rare cases of familial meningiomas without underlying genetic syndrome have been reported [12]. Meningioangiomatosis (MA) has the most notable association with meningioma and is further discussed below.
Neurofibromatosis type 2
After vestibular schwannoma, meningiomas are the second most frequent tumor in patients with NF2, seen in 53% of patients and 83% of those with the early onset, more severe, “Wishart variant” [13]. However, neurofibromatosis type 1 is not associated with an increased risk of meningioma [14]. NF2-associated meningiomas have not been shown to differ significantly from sporadic pediatric tumors with regard to clinicopathological and genotypic features. However, NF2-associated meningiomas have a higher frequency of multiplicity and merlin loss, and sporadic pediatric meningiomas have a higher incidence of brain invasion [15]. Patients with NF2 have a higher risk of developing meningiomas of the spinal canal and optic nerve sheath [16]. Loss of NF2 gene expression (which is located on 22q12) occurs in almost all NF2-associated meningiomas and 40–60% of sporadic meningiomas [17]. The high incidence of meningiomas in NF2, the fact that this population is more prone to develop multiple meningiomas, and presentation at an earlier age conforms to Knudson’s “two-hit” hypothesis, in that only one additional hit of the NF2 gene is required for tumorigenesis.
Ionizing radiation
Cranial radiotherapy is a risk factor for meningioma development. There is a nearly tenfold higher risk for children with radiation exposure compared to those without [18]. The high sensitivity of arachnoidal tissue to irradiation in the meninges of children increases vulnerability to oncogenic stimulation. Ionizing radiation causes mutations in the genome, either directly or indirectly via formation of free radicals. High doses elicit a more rapid loss of cellular control mechanisms and failure of the DNA repair system, eventually leading to tumor formation. Radiation-induced meningiomas rarely manifest in childhood, although they remain an ever-increasing problem among adults, allowing for the latency period from the time of irradiation to clinical onset and that the incidence of meningiomas is known to increase as a function of time [19].
A comprehensive literature review of radiation-induced brain tumors after CNS irradiation in childhood has been recently published [20]. Primary diseases encompassed both CNS and hematological tumors. Thirty-three World Health Organization (WHO) grade I and seven WHO grade II meningiomas of atypical subtype were identified. There was no clear correlation between dose of radiotherapy delivered and histological grade of the secondary meningioma. The latency period for the occurrence of secondary atypical meningiomas did not differ between children and adults, whereas the mean latency period for the occurrence of secondary benign meningiomas was longer in adults.
Compared with spontaneous meningiomas, radiation-induced meningiomas in adults are characterized by younger age at diagnosis, atypical or malignant pathology, multiplicity, and recurrence [21]. In children, radiation-induced meningiomas arise within the irradiation field by definition, are more likely to be multifocal, and have high proliferative potential and high degrees of atypia and mitosis. Higher levels of radiation exposure are associated with a shorter latency period, suggesting a dose effect [18]. The latency period is shorter for patients irradiated at less than five years of age and for those who developed atypical or anaplastic meningiomas [5].
Radiation-induced meningiomas often have complex structural and numerical chromosomal abnormalities consistent with DNA damage induced by irradiation. Loss of 1p, 6q, 7p, and chromosome 22 have been described in adult radiation-induced meningiomas [22, 23]. NF2 gene alterations play a less important role in the pathogenesis of radiation-induced meningioma than sporadic meningiomas without previous history of radiation [24]. No specific molecular patterns enabling differentiation between radio-induced and spontaneous lesions have been identified. Specific cytogenetic signatures of pediatric radiation-induced meningiomas have yet to be ascertained. Brassesco et al. [25] have demonstrated loss of 1p and complex aberrations of chromosomes 1, 6, and 12 in a pediatric radiation-induced meningioma.
Meningioangiomatosis
MA is a rare hamartomatous lesion of the CNS characterized by proliferation of meningothelial cells, microvasculature, and fibroblasts. It can occur sporadically or in the setting of neurofibromatosis (NF), especially type 2. Patients can be asymptomatic, especially those with NF, or present with a long history of seizures and/or headache, as is often seen in sporadic cases. Meningioma associated with MA is even rarer. The total number of cases reported under the age of 18 of meningioma associated with MA is 19 [4, 26–31]. There is a male predominance (12 males, 7 females), with all cases located in the fronto-temporal cortex. All meningiomas were WHO grade I tumors except for one atypical meningioma. One case had NF. The pathogenesis of this condition remains unclear. Cytogenetic studies have demonstrated loss of NF2 gene expression in both the MA and meningioma components but a much higher proliferation index (MIB-1 labeling index (LI)) in the meningioma than in the MA component. This suggests that both the meningioma and MA undergo the same overlapping clonal process with the meningioma undergoing additional genetic alterations that confer a greater proliferative potential [29, 32, 33]. In benign meningiomas associated with MA, histological appearances are known to mimic brain invasion, so careful histopathological interpretation is necessary. Outcomes in this subgroup have generally been favorable. Five deaths have been recorded—two postoperative, one disease-related, one unspecified, and one unrelated. Thirteen patients were alive at the end of their follow-up periods, with one having sustained a recurrence two years after subtotal resection of the tumor. This patient had a high MIB-1 LI of 15%. The recurrence was treated with stereotactic radiosurgery and no further recurrence occurred during the four-year follow-up [31]. Clinical outcome was unspecified for one patient.
Clinical features
Common presenting signs and symptoms include those of raised intracranial pressure (headache, vomiting and papilledema), seizures, visual disturbance, motor disturbance, and cranial nerve defects. The most frequently affected CNS locations in children are supratentorial and include the cerebral convexity, intraventricular, anterior and middle cranial fossae, falcine and parasagittal regions. Infratentorial meningiomas occur more frequently in children than in adults (20% [26] vs. 8–10% [34]). There is a greater incidence of intraventricular meningiomas in children (11.3% [26] vs. 0.5–2% [35]). Of intraventricular tumors, third ventricular tumors are rare [36]. Meningiomas can occur in other unusual locations in children, for example intraparenchymal, intraosseus and deep within the Sylvian fissure. One adult study has reported distinct patterns of gene expression in spinal meningiomas compared with intracranial meningiomas [37], suggesting spinal meningiomas are a different clinical, biological, and genetic entity, although no studies have been subsequently performed.
Histological and immunohistochemical features
Meningiomas are histologically classified according to the WHO grading system. The most recent revision of the WHO grading system was published in 2007, with the most significant change recognizing brain invasion as reflecting more aggressive biological potential. Most pediatric meningiomas are WHO grade I (80.6%) with WHO grade II (10.4%) and III (8.1%) tumors occurring less frequently (Figs. 1, 2 and 3) [38].

Sheets of tumor cells with uniform oval nuclei from a meningioma, WHO grade I

Atypical meningioma with brain invasion, WHO grade II

Marked nuclear atypia and elevated mitotic index (>20 mitotic figures/10 high-power fields) in an anaplastic meningioma, WHO grade III
In adults, the incidence of WHO grade II and III meningiomas is lower than in children, constituting 4.7–7.2% and 1.0–2.8% of cases, respectively [39]. Metastatic meningioma is extremely rare in children, with isolated cases of extracranial metastases reported in the lung, liver, lymph nodes, and bone in patients with WHO grade III meningiomas [26, 40–42]. Similarly, metastatic disease is rare in adults, estimated to occur in 0.1% of patients [43]. There is a known association between clinical behavior and histological grade. The association is stronger for sporadic adult meningiomas, because one study has shown recurrence/death to be relatively common in pediatric patients with WHO grade I tumors [15]. However, recurrence-free survival was significantly related to WHO grade in the largest published cohort of pediatric meningiomas [9]. Atypical and malignant transformation is a recognized phenomenon and has been identified in several cases at recurrence [44].
In adults, the staining index for the anti-Ki-67 monoclonal antibody, MIB-1, is correlated with histological atypia and tumor recurrence [45–50]. Ki-67 is a nuclear antigen expressed during active phases of the cell cycle (G1, S, G2, and M phases) but is absent during the G0 phase. The monoclonal antibody, MIB-1, detects the Ki-67 antigen and is used as a marker of cellular proliferation. A higher MIB-1 LI indicates shorter cell cycle times and faster tumor growth [50]. In pediatric meningiomas, most studies concur that MIB-1 LI correlates with tumor grade [15, 27, 50], although there is substantial overlap of MIB-1 LI values between histological grades and significant inter-institutional variation is often noted because of different counting and staining techniques. One study correlated elevated MIB-1 LI with meningioma recurrence [50], whereas other studies did not demonstrate statistical significance [1, 15, 27, 51]. Im et al. [4] correlated high MIB-1 LI with large tumor size and short symptom duration, but not with adverse outcome or higher histological grade. Although MIB-1 LI provides a useful adjunct in relating tumor to histological grade in children, it cannot be used in isolation because of substantial overlap between histological grades. There is a paucity of data to support its use in predicting recurrence in pediatric meningioma. Other immunohistochemical markers that are useful for confirming meningothelial phenotype include epithelial membrane antigen and vimentin.
Based on an association with breast cancer and female sex predominance in combination with observations of acceleration of tumor growth during pregnancy and the luteal phase of the menstrual cycle in adults, a tumorigenic role of hormones in meningioma has been suggested. Adult hormone immunohistochemical receptor studies revealed an inverse relationship between progesterone receptor (PR) expression and tumor proliferation and histological grade [17, 52]. In adult meningiomas, gene expression has revealed an association with PR status, with significant up-regulation of genes on the long arm of chromosome 22 and near the NF2 gene in PR positive lesions compared with PR-negative lesions, suggesting a higher rate of 22q loss in PR-negative lesions [53]. A clear consensus on the prognostic value of PR status in predicting recurrence in adult meningioma is yet to be drawn. Some studies have suggested PR negativity as a strong predictor for recurrence, whereas others have been unable to find a statistically significant correlation between the two variables [54–56]. Immunohistochemical expression of the estrogen receptor (ER) is traditionally low in adult meningioma, and its role is less well described. A few recent reports have demonstrated higher ER expression in meningioma, although not to the levels observed for PR expression [52, 57]. ER-positive tumors have been correlated with aggressive clinical behavior, higher proliferation indices, progression, recurrence, and karyotype abnormalities in a small number of recent studies, although much more validation is required before it can be regarded as an adjunctive prognostic variable [58, 59]. Androgen, somatostatin, growth hormone, and prolactin receptors have also been identified in adult meningiomas.
There is scant information about children but reports tend to concur with adult data, that PRs are more frequently present than ERs. Sheikh et al. [60] found three of their WHO grade I specimens negative for ER and positive for PR. In keeping with adult meningioma, Perry et al. [17] observed an approximate inverse association between PR expression and tumor grade in pediatric meningiomas. PR expression was of similar frequency, irrespective of age at presentation [17, 61]. PR status was not related to clinical outcome. PR status as an adjunctive diagnostic marker in pediatric meningioma is limited because of the degree of overlap amongst histological grades and because both PR positivity and negativity are encountered in all grades. The role of hormones and their receptors in the pathogenesis of meningiomas remains to be elucidated.
In adult meningioma, the prognostic value of p53 immunohistochemical overexpression remains controversial. Although most agree that higher p53 expression correlates with poorer histological grade, there is no clear consensus as to whether there is an association with recurrence [1, 45, 46]. p53 immunohistochemistry in pediatric meningioma is not well described. For twenty-seven tumors Gao et al. [1] could not relate p53 immunoreactivity to recurrence. In addition, p53 expression in four tumors described by Alexiou et al. [51] did not relate to histological grade or outcome.
Interestingly, a recent report of an 11-month-old child, in whom there was growth of chronic bilateral subdural collections, demonstrated overexpression of aquaporin 1 in the meningioma specimen and blood vessels on immunohistochemical staining. This suggests strong expression of aquaporin 1 in tumor vessels could be linked to cyst formation and fluid overproduction leading to subdural collections [62]. This finding has yet to be repeated.
Radiological features
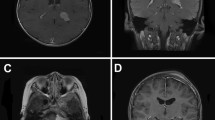
On magnetic resonance imaging (MRI), meningiomas are iso-hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging. Marked gadolinium enhancement is seen (Fig. 4). MRI is superior to computed tomography (CT) in providing detailed information about the tumor. MRI enables better delineation of tumor borders, associated edema, interfaces, and cystic areas. The “dural tail sign” can occur as a characteristic feature, although it is not present in every case, and reflects neoplastic dural infiltration or reactive vascularity or both, draining into the adjacent dura. CT appearances are of a hyperdense mass with variable calcification, edema, and hyperostosis. CT is more effective than MRI at demonstrating the effects on adjacent bone [63]. On CT, meningiomas enhance homogeneously and diffusely after contrast [60, 64–66]. Several studies have looked at the correlation between MRI signal changes, diffusion-weighted MR, MR spectroscopy, and positron emission tomography with histopathology in adult meningiomas. Whereas some studies have revealed a correlation between imaging features and tumor grade, others have not. Consequently, no clear consensus has been reached [63, 67–73].

MRI demonstrating a vividly enhancing solid meningioma in the superior aspect of the right cerebral hemisphere associated with non-wall enhancing cysts. Cyst indicated by arrow
Meningiomas can also have a cystic component (Fig. 4). Pediatric meningiomas are more likely to be cystic than their adult counterparts (24% vs. 2–4%) [74]. Four types of cyst have been described in meningiomas. The cyst may be intra-tumoral, either in the center (type I) or on the periphery (type II), or peri-tumoral, either within the brain substance (type III) or at the brain-tumor interface (type IV) [60]. Type II cysts are the most frequent in pediatric meningiomas [75]. Cysts form after necrosis, degeneration, and hemorrhage. Glial response or arachnoid reaction may also contribute [1]. Pediatric meningiomas have been noted to be larger in size compared to adult tumors [64]. Multiple meningioma at presentation is less common in children than adults (2.5% vs. 8–9%) [76]. Lack of dural attachment is rare in adults but can be found in up to 13–30% of childhood cases [77]. Calcification is another finding that can be present on imaging (Fig. 5) and is more readily seen on CT than MRI.

MRI demonstrating punctate foci of calcification seen within a solid meningioma in the superior aspect of the right cerebral hemisphere associated with marked peri-tumoral and peri-cystic edema. Peri-tumoral edema indicated by arrow
Molecular abnormalities
In adults, meningioma was the first solid tumor associated with a characteristic cytogenetic abnormality, monosomy 22 [78, 79]. Subsequent studies revealed that loss of NF2 gene expression was an early tumorigenic event. Loss of heterozygosity on chromosome 22q with bi-allelic inactivation of the NF2 tumor suppressor gene is one of the most frequently observed genetic alterations [17]. Merlin is the resulting protein product of NF2 gene expression and is thought to play a role in regulating cell growth (particularly under conditions of increased cell density), cell motility, binding of several transmembrane signaling proteins, and interaction with important proteins. Meningioma-associated NF2 mutations result in a truncated non-functional merlin protein. Other genes located on chromosome 22q that have been implicated in meningioma tumorigenesis include BAM22, breakpoint cluster region (BCR), and TIMP metallopeptidase inhibitor 1 (TIMP-1), the latter in higher-grade meningiomas [43]. Protein 4.1 molecules are structurally similar to merlin and losses of 4.1R and 4.1B protein, because of differentially expressed in adenocarcinoma of the lung 1 (DAL1) gene deletions on chromosome 18p11.32, have been demonstrated in adult meningiomas. The precise functions of protein 4.1 tumor suppressor genes have yet to be elucidated. Deleted in liver cancer 1 (DLC1) is a recently identified tumor suppressor gene located on chromosome 8p21.3-22 that may have a role in meningioma formation [80].
In adults, cytogenetic alterations associated with meningioma progression and atypical/anaplastic histology are well characterized. These include the presence of dicentric or ring chromosomes, losses of chromosome arms 1p, 6q, 9p, 10, 14q, and 18q, and gains/amplifications on 1q, 9q, 12q, 15q, 17q, and 20q [81]. Alterations of the cyclin D-dependent kinase tumor suppressor genes, CDK2NA (p16 INK4a, p14 ARF) and CDK2NB (p15 INK4b), on chromosome 9p21 are found in approximately two thirds of anaplastic meningiomas [82]. Loss of cell adhesion molecule 1 (CADM1) gene expression, previously known as tumor suppressor in lung cancer-1 (TSLC1), on chromosome 11q23 has been correlated with increased tumor grade [83]. Aberrant CpG island hypermethylation has been associated with atypical and anaplastic meningiomas [84]. Whereas NF2 and DAL1 deletions are strongly implicated in meningioma pathogenesis, 1p and 14q deletions are progression-associated markers associated with higher tumor grade. Reduced expression of maternally expressed gene 3 (MEG3) and N-myc downstream regulated gene 2 (NDRG2) have been identified on chromosome 14q [43].
Overexpression of various growth factors, for example platelet-derived growth factor (PDGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), insulin like growth factor (IGF), and transforming growth factor-beta (TGF-β), and their receptors, together with their downstream signaling pathways, including Ras/mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT, have been shown to occur in the pathogenesis of adult meningioma [85]. Deregulation of the Notch, hedgehog, and beta-catenin/wnt pathways have also been identified [86].
Limited molecular characterization has been performed in pediatric meningiomas. Most reports consist of only a few cases. As with adults, chromosome 22 abnormalities are the most frequent change [87–91]. Biegel et al. [87] also described ring and dicentric chromosomes, loss of chromosome 6, and loss of chromosome 1 in several cases in their initial series and suggested that complicated karyotypes may be associated with increased risk of recurrence. Perilongo et al. [65] performed karyotype analysis for five patients. Three had chromosome 22 abnormalities, with two having more complex numerical and structural changes. Zwerdling and Dothage [92] performed karyotype analysis on eight patients; all yielded abnormal results. Three had chromosome 22 abnormalities. Several other unusual trisomies, translocations, duplications, and deletions were also observed. Perry et al. [15] found that co-deletion of four chromosome sites (NF2, DAL1, 1p, and 14q) increased with tumor grade. NF2 and DAL1 deletions, with corresponding loss of protein products, were common across all tumor grades, further supporting the role of these genetic lesions in early tumor development. 1p and 14q deletions, both individually and in combination, were more frequently observed in higher tumor grades. The association was weaker than reported in adult meningiomas, because these deletions were also frequently evident in histologically benign pediatric meningiomas. Although pediatric and NF2-associated meningiomas have been shown to share common molecular alterations with their adult sporadic counterparts, a higher fraction are genotypically and phenotypically more aggressive [15].
Treatment options
The prognosis of pediatric meningioma has improved over the years with advances in surgical techniques and supportive care. The largest case series of 87 pediatric meningiomas reported a mortality of 10.6% and recurrence of 19.4%. Other than recurrence-free survival being significantly related to WHO grade in this cohort [9], there has been no statistical validation of prognostic factors and treatment of pediatric meningioma. Recommendations have largely been drawn from retrospective case analyses and the treatment of adult meningioma.
Gross total resection is the ultimate therapeutic objective in children [16] and adults, and re-resection should be considered in the event of initial subtotal resection. Resection of the dural origin/attachment is recommended, because there is a higher reported incidence of recurrence if dural attachment is left behind [16]. In children, tumor size, vascularity, and location may hamper the ability to perform a gross total resection, because of the risk of excessive intra-operative hemorrhage and post-operative neurological morbidity. In this instance, staged multiple procedures are recommended [16]. Pre-operative embolization may be useful in giant vascular tumors, although the risk of procedural morbidity because of small vessel caliber in children must be taken into account.
Because of lack of evidence for children and adults, guidelines for radiotherapy are limited, and use varies significantly between institutions. Recommendations are based on adult retrospective series [16, 93]. Current National Institute for Health and Clinical Excellence recommendations for adult patients suggest radiotherapy be considered for those with WHO grade II or III tumors, multiple relapses, contraindication to surgery, invasion of adjacent brain, or extensive invasion of other tissues [94]. The National Cancer Institute (NCI) is currently conducting a phase II risk stratified trial of observation or radiotherapy in adults with WHO grade I, II, or III meningioma (http://www.cancer.gov). In children, the use of radiotherapy must be balanced against the risk of significant late sequelae. This is particularly important in infants and young children, whose CNS is more vulnerable to the effects of radiotherapy. The Children’s Cancer and Leukaemia Group (CCLG) suggests consideration of three-dimensional (3D), conventionally fractionated, conformal radiotherapy in WHO grade I and II meningiomas after multiple relapses not amenable to further surgery or evidence of clinically relevant progression after incomplete resection, especially if the tumor threatens to compromise vital functions, and in all WHO grade III meningiomas at time of diagnosis, irrespective of surgical outcome [16]. The use of stereotactic radiosurgery has been reported for adults with small tumors [16, 39, 95], however there are limited data on the use of this modality in children. Stereotactic radiosurgery was used for successful treatment of a child with recurrence of meningioma in association with MA [31]. Gamma-knife radiosurgery was successful for three children with recurrent meningioma [4, 96]. Use of proton beam therapy and intensity-modulated radiation therapy in adults has had promising results [39, 82, 97], although use is limited as these modalities are not currently widely available compared with 3D conformational radiotherapy.
Use of drug therapy in the treatment of meningioma has been disappointing. In adult meningioma, various chemotherapeutic agents have been investigated, including single-agent temozolomide and irrinotecan, and combinations of adriamycin, vincristine, dacarbazine, and cyclophosphamide, or high-dose ifosfamide. However, none has demonstrated any benefit [85]. Hydroxyurea, an oral ribonucleotide reductase inhibitor, was marginally beneficial. Stabilization of progressive adult meningiomas using interferon alpha-2B has been reported in a small cohort of patients. Anti-hormonal therapies have not been successful using mifepristone, an anti-progesterone agent, or tamoxifen, an anti-estrogen agent [85].
Trials of molecularly targeted therapies have included the EGF receptor inhibitors, gefitinib and erlotinib. However, they did not have significant activity against recurrent meningioma [85]. Several NCI phase II trials are currently open, for recurrent or progressive adult meningiomas, investigating a variety of novel molecularly targeted therapies including bevacizumab (VEGF antibody) alone and in combination with everolimus (mammalian target of rapamycin (mTOR) inhibitor), hydroxyurea, and imatinib mesylate (PDGF receptor inhibitor), vatalanib (VEGF receptor and PDGF receptor inhibitor), and sunitinib malate (VEGF receptor and PDGF receptor inhibitor) (http://www.cancer.gov).
In-vivo experiments have demonstrated growth-inhibitory effects of several different agents including calcium channel antagonists [86]. Consequently, verapamil, a calcium channel antagonist, is currently being investigated in a NCI phase II trial, in combination with hydroxyurea for recurrent or progressive adult meningiomas (http://www.cancer.gov). A pilot study using the sustained-release somatostatin receptor agonist, sandostatin LAR, has revealed some efficacy against meningiomas which overexpressed the somatostatin receptor [85]. Pasireotide LAR, a somatostatin receptor agonist, is being investigated in a NCI phase II trial for recurrent or progressive adult meningiomas (http://www.cancer.gov).
Use of chemotherapy in eight pediatric meningioma cases has been reported. One patient remained alive three years after receiving hydroxyurea and an unspecified phase II agent after subtotal resection of a malignant meningioma [92]. Another patient remained alive four years after gross total resection of a rhabdoid meningioma and receipt of unspecified chemotherapy (J. Sudhir, personal communication). Another case received unspecified chemotherapy and was lost to follow-up [9]. The other five patients died—in three chemotherapy was unspecified [9, 42, 98], one received intrathecal methotraxate and radiation after gross total resection of a meningothelial meningioma, and the other received vincristine, ifosfamide, adriamycin, and cyclophosphamide and radiation after subtotal resection of a malignant meningioma [92]. There are no pediatric-specific meningioma drug trials currently open. Based on current evidence there is no recommendation for the use of drug therapy in children or adults with meningioma.
In a pediatric patient in whom meningioma is diagnosed, work up should include assessment for presence of NF2 and Gorlin syndrome. Referral to a geneticist should be undertaken, because manifestations of NF2 are often absent at the time of presentation and somatic mosaicism can occur in up to 33% of patients with NF2, which may explain milder phenotypes. New germline mutations account for 50% of NF cases and so family history is frequently negative [14]. Life-table analyses from adult series have revealed relapses as late as 15 years after initial surgery, so long-term follow-up for pediatric patients is mandatory [92]. The CCLG recommend annual MRI surveillance for at least 10 years [16]. In the setting of NF2, Gorlin syndrome, or a previous history of cranial irradiation, follow-up should be life-long. Long term sequelae of pediatric meningioma survivors should not be overlooked and include visual deficits, neuropsychological abnormalities, developmental delay, CNS palsies, motor deficits, seizure disorders, and sensory deficits.
Future directions
We have entered an era of technology that enables screening of genes at the molecular level to characterize the molecular genetic signatures of tumors. Gene expression profiling has been undertaken in meningiomas derived from adult patients and has enabled the identification of specific deregulated pathways, novel genes implicated in meningioma pathogenesis, and detailed characterization of gene expression according to tumor grade and gender. SNP-array karyotyping is enabling identification of a higher frequency of genomic lesions than traditional cytogenetics, and microRNA profiling is opening up new avenues for exploration.
An extensive body of molecular biology data exists for several pediatric CNS tumors, most notably ependymoma, medulloblastoma, and glioma. These data have enabled identification of deregulated growth factors, receptors, and signaling pathways which are potential targets for pharmacological intervention. In addition, several molecular markers have been identified to assist in guiding diagnosis and prognosis. However, genome-wide profiling of pediatric meningioma has not yet been undertaken. The number of striking differences between adult and pediatric meningiomas probably reflects differing biology, as with other pediatric CNS tumors, and so treatment considerations and the molecular characteristics of pediatric meningiomas should not be presumed to be the same. Within the current literature, statistical validation regarding prognostic variables and treatment for pediatric meningioma is lacking. We must take advantage of these sophisticated molecular biology tools to molecularly characterize pediatric meningiomas, and use the knowledge in the clinical setting, such that a therapeutic international prospective randomized controlled trial, which includes collection of tumor samples, can be considered in the not too distant future. Only then are we likely to achieve significant progress in the treatment of pediatric meningioma.
References
Gao X, Zhang R, Mao Y, Wang Y (2009) Childhood and juvenile meningiomas. Childs Nerv Syst 25(12):1571–1580
Claus EB, Bondy ML, Schildkraut JM, Wiemels JL, Wrensch M, Black PM (2005) Epidemiology of intracranial meningioma. Neurosurgery 57:1088–1095
Turgut M, Ozcan OE, Bertan V (1997) Meningiomas in childhood and adolescence: a report of 13 cases and review of the literature. Br J Neurosurg 11(6):501–507
Im SH, Wang KC, Kim SK, Oh CW, Kim DG, Hong SK, Kim NR, Chi JG, Cho BK (2001) Childhood meningioma: unusual location, atypical radiological findings and favorable treatment outcome. Childs Nerv Syst 17:656–662
Perry A, Dehner LP (2003) Meningeal tumors of childhood and infancy. An update and literature review. Brain Pathol 13:386–408
CBTRUS (2010) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2004–2006. Central Brain Tumor Registry of the United States, Hinsdale
Albrecht S, Goodman JC, Rajagopolan S, Levy M, Cech DA, Cooley LD (1994) Malignant meningioma in Gorlin’s syndrome: cytogenetic and p53 gene analysis. Case report. J Neurosurg 81:466–471
Kimonis VE, Mehta SG, Digiovanna JJ, Bale SJ, Pastakia B (2004) Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med 6:495–502
Rushing EJ, Olsen C, Mena H, Rueda ME, Lee YS, Keating RF, Packer RJ, Santi M (2005) Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg 103(6 Suppl):489–495
Kepes JJ, Chen WY, Connors MH, Vogel FS (1988) “Chordoid” meningeal tumors in young individuals with peritumoral lymphoplasmacellular infiltrates causing systemic manifestations of the Castleman syndrome. A report of seven cases. Cancer 62:391–406
Miller RW, Rubinstein LJ (1995) Tumors in Rubenstein–Taybi syndrome. Am J Med Genet 56:112–115
Sieb JP, Puist S-M, Buch A (1992) Familial CNS tumours. J Neurol 239:343–344
Evans DGR, Birch JM, Ramsden RT (1999) Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child 81:496–499
Goutagny S, Kalamarides M (2010) Meningiomas and neurofibromatosis. J Neurooncol 99:341–347
Perry A, Giannini C, Raghavan R, Scheithauer BW, Banerjee R, Margraf L, Bowers DC, Lytle RA, Newsham IF, Gutmann DH (2001) Aggressive phenotypic and genotypic features in pediatric and NF2-associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol 60(10):994–1003
Traunecker H, Mallucci C, Grundy R, Pizer B, Saran F (2008) Children’s cancer and leukaemia group (CCLG): guidelines for the management of intracranial meningioma in children and young people. Br J Neurosurg 22(1):13–25
Perry A, Gutmann DH, Reifenberger G (2004) Molecular pathogenesis of meningiomas. J Neurooncol 70:183–202
Menon G, Nair S, Sudhir J, Rao BR, Mathew A, Bahuleyan B (2009) Childhood and adolescent meningiomas: a report of 38 cases and review of the literature. Acta Neurochir 151:239–244
Banerjee J, Paakko E, Harila M, Herva R, Tuominen J, Koivula A, Lanning M, Harila-Saari A (2009) Radiation-induced meningiomas: a shadow in the success story of childhood leukaemia. Neuro Oncol 11:543–549
Pettorini BL, Park YS, Caldarelli M, Massimi L, Tamburrini G, Di Rocco C (2008) Radiation-induced brain tumours after central nervous system irradiation in childhood: a review. Childs Nerv Syst 24:793–805
Greene S, Nair N, Ojemann JG, Ellenbogen RG, Avellino AM (2008) Meningiomas in children. Pediatr Neurosurg 44:9–13
Lillehei KO, Donson AM, Kleinschmidt-DeMasters BK (2008) Radiation-induced meningiomas: clinical, cytogenetic and microarray features. Acta Neuropathol 116:289–301
Rajcan-Separovic E, Maguire J, Loukianova T, Nisha M, Kalousek D (2003) Loss of 1p and 7p in radiation-induced meningiomas identified by comparative genomic hybridization. Cancer Genet Cytogenet 144:6–11
Joachim T, Ram Z, Rappaport ZH, Simon M, Schramm J, Wiestler OD, von Deimling A (2001) Comparative analysis of the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and radiation-induced meningiomas. Int J Cancer 94:218–221
Brassesco MS, Valera ET, Neder L, Castro-Gamero AM, de Oliveira FM, Santos AC, Scrideli CA, Oliveira RS, Machado HR, Tone LG (2009) Childhood radiation-associated atypical meningioma with novel complex rearrangements involving chromosomes 1 and 12. Neuropathology 29:585–590
Choux M, Lena G, Genitoro L, Azambuja N, Cunha C, Dechambenoit G (1991) Meningiomas in children. In: Schmidek HH (ed) Meningiomas and their surgical management. WB Saunders, Philadelphia, pp 93–102
Demirtas E, Ersahin Y, Yilmaz F, Mutluer S, Veral A (2000) Intracranial meningeal tumours in childhood: a clinicopathologic study including MIB-1 immunohistochemistry. Pathol Res Pract 196:151–158
Deb P, Gupta A, Sharma MC, Gaikwad S, Singh VP, Sarkar C (2006) Meningioangiomatosis with meningioma: an uncommon association of a rare entity—report of a case and review of the literature. Childs Nerv Syst 22:78–83
Kim NR, Cho SJ, Suh YL (2009) Allelic loss on chromosomes 1p32, 9p21, 13q14, 16q22, 17p and 22q12 in meningiomas associated with meningioangiomatosis and pure meningioangiomatosis. J Neurooncol 94:425–430
Saad A, Folkerth R, Poussaint T, Smith E, Ligon K (2009) Meningioangiomatosis associated with meningioma: a case report. Acta Cytol 53:93–97
Kim NR, Choe G, Shin SH, Wang KC, Cho BK, Choi KS, Chi JG (2002) Childhood meningiomas associated with meningioangiomatosis: report of five cases and literature review. Neuropathol Appl Neurobiol 28:48–56
Sinkre P, Perry A, Cai D, Raghavan R, Watson M, Wilson K, Barton Rogers B (2001) Deletion of the NF2 region in both meningioma and juxtaposed meningioangiomatosis: case report supporting a neoplastic relationship. Pediatr Dev Pathol 4:568–572
Perry A, Kurtkaya-Yapicier O, Scheithauer BW, Robinson S, Prayson RA, Kleinschmidt-DeMasters BK, Stemmer-Rachamimov AO, Gutmann DH (2005) Insights into meningioangiomatosis with and without meningioma: a clinicopathologic and genetic series of 24 cases with review of the literature. Brain Pathol 15(1):55–65
Di Rocco C, Di Rienzo A (1999) Meningiomas in childhood. Crit Rev Neurosurg 9:180–188
Liu M, Wei Y, Liu Y, Zhu S, Li X (2006) Intraventricular meningiomas: a report of 25 cases. Neurosurg Rev 29:36–40
Martinez-Lage JF, Poza M, Alcaraz J, Molina E (1993) Giant meningioma of the III ventricle in a child: a case report and review of the literature. Childs Nerv Syst 9:306–308
Sayagues JM, Tabernero MD, Maillo A, Trelles O, Espinosa AB, Sarasquete ME, Merino M, Rasillo A, Vera JF, Santos-Briz A, de Alava E, Garcia-Macias MC, Orfao A (2006) Microarray-based analysis of spinal versus intracranial meningiomas: different clinical, biological and genetic characteristics associated with distinct patterns of gene expression. J Neuropathol Exp Neurol 65(5):445–454
Kotecha RS (2010) In: Proceedings of the 24th annual scientific meeting of the Australian and New Zealand Children’s Haematology and Oncology Group, Sydney, Australia
Modha A, Gutin PH (2005) Diagnosis and treatment of atypical and anaplastic meningiomas: a review. Neurosurgery 57:538–550
Enam SA, Abdulrauf S, Mehta B, Malik GM, Mahmood A (1996) Metastasis in meningioma. Acta Neurochir (Wien) 138:1172–1177
Doxtader EE, Butts SC, Holsapple JW, Fuller CE (2009) Aggressive pediatric meningioma with soft tissue and lymph node metastases: a case report. Pediatr Dev Pathol 12(3):244–248
Baumgartner JE, Sorenson JM (1996) Meningioma in the pediatric population. J Neurooncol 29:223–228
Mawrin C, Perry A (2010) Pathological classification and molecular genetics of meningiomas. J Neurooncol 99:379–391
Arivazhagan A, Devi BI, Kolluri SV, Abraham RG, Sampath S, Chandramouli BA (2008) Pediatric intracranial meningiomas—do they differ from their counterparts in adults? Pediatr Neurosurg 44:43–48
Terzi A, Saglam EA, Barak A, Soylemezoglu F (2008) The significance of immunohistochemical expression of Ki-67, p53, p21 and p16 in meningiomas tissue arrays. Pathol Res Pract 204(5):305–314
Ozen O, Demirhan B, Altinors N (2005) Correlation between histological grade and MIB-1 and p53 immunoreactivity in meningiomas. Clin Neuropathol 24(5):219–224
Bruna J, Brell M, Ferrer I, Gimenez-Bonafe P, Tortosa A (2007) Ki-67 proliferative index predicts clinical outcome in patients with atypical or anaplastic meningioma. Neuropathology 27:114–120
Torp SH, Lindboe CF, Gronberg BH, Lydersen S, Sundstrom S (2005) Prognostic significance of Ki-67/MIB-1 proliferation index in meningiomas. Clin Neuropathol 24(4):170–174
Uzum N, Ataoglu GA (2008) Histopathological parameters with Ki-67 and bcl-2 in the prognosis of meningiomas according to WHO 2000 classification. Tumori 94(3):389–397
Sandberg DI, Edgar MA, Resch L, Rutka JT, Becker LE, Souweidane MM (2001) MIB-1 staining index of pediatric meningiomas. Neurosurgery 48:590–597
Alexiou GA, Mpairamidis E, Psarros A, Sfakianos G, Prodromou N (2008) Intracranial meningiomas in children: report of 8 cases. Pediatr Neurosurg 44:373–375
Omulecka A, Papierz W, Nawrocka-Kunecka A, Lewy-Trenda I (2006) Immunohistochemical expression of progesterone and estrogen receptors in meningiomas. Folia Neuropathol 44(2):111–115
Claus EB, Park PJ, Carroll R, Chan J, Black PM (2008) Specific genes expressed in association with progesterone receptors in meningioma. Cancer Res 68(1):314–322
Maiuri F, De Caro Mdel B, Esposito F, Cappabianca P, Strazzullo V, Pettinato G, de Divitiis E (2007) Recurrences of meningiomas: predictive value of pathological features and hormonal and growth factors. J Neurooncol 82(1):63–68
Roser F, Nakamura M, Bellinzona M, Rosahl SK, Ostertag H, Samii M (2004) The prognostic value of progesterone receptor status in meningiomas. J Clin Pathol 57(10):1033–1037
Guevara P, Escobar-Arriaga E, Saavedra-Perez D, Martinez-Rumayor A, Flores-Estrada D, Rembao D, Calderon A, Sotelo J, Arrieta O (2010) Angiogenesis and expression of estrogen and progesterone receptors as predictive factors for recurrence of meningioma. J Neurooncol 98(3):379–384
Leaes CG, Meurer RT, Coutinho LB, Ferreira NP, Pereira-Lima JF, da Costa Oliveira M (2010) Immunohistochemical expression of aromatase and estrogen, androgen and progesterone receptors in normal and neoplastic human meningeal cells. Neuropathology 30(1):44–49
Pravdenkova S, Al-Mefty O, Sawyer J, Husain M (2006) Progesterone and estrogen receptors: opposing prognostic indicators in meningiomas. J Neurosurg 105(2):163–173
Korhonen K, Salminen T, Raitanen J, Auvinen A, Isola J, Haapasalo H (2006) Female predominance in meningiomas can not be explained by differences in progesterone, estrogen or androgen receptor expression. J Neurooncol 80(1):1–7
Sheikh BY, Siqueira E, Dayel F (1996) Meningioma in children: a report of nine cases and review of the literature. Surg Neurol 45:328–335
Roser F, Nakamura M, Ritz R, Bellinzona M, Dietz K, Samii M, Tatagiba MS (2005) Proliferation and progesterone receptor status in benign meningiomas are not age dependent. Cancer 104(3):598–601
Marton E, Feletti A, Basaldella L, Des Tos AP, Bendini M, Longatti P (2008) Atypical cystic meningioma overexpressing AQP1 in early infancy; case report with literature review. Acta Paediatr 97:1145–1149
Saloner D, Uzelac A, Hetts S, Martin A, Dillon W (2010) Modern meningioma imaging techniques. J Neurooncol 99:333–340
Darling CF, Byrd SE, Reyes-Mugica M, Tomita T, Osborn RE, Radkowski MA, Allen ED (1994) MR of pediatric intracranial meningiomas. Am J Neuroradiol 15:435–444
Perilongo G, Sutton LN, Goldwein JW, Gusnard D, Schut L, Biegel JA, Rorke LB, Lange B, D’Angio GJ (1992) Childhood meningiomas: experience in the modern imaging era. Pediatr Neurosurg 18:16–23
Glasier CM, Husain MM, Chadduck W, Boop FA (1993) Meningiomas in children: MR and histopathologic findings. Am J Neuroradiol 14:237–241
Monleon D, Morales JM, Gonzalez-Darder J, Talamantes F, Cortés O, Gil-Benso R, López-Ginés C, Cerdá-Nicolás M, Celda B (2008) Benign and atypical meningioma metabolic signatures by high-resolution magic-angle spinning molecular profiling. J Proteome Res 7(7):2882–2888
Hakyemez B, Yildirim N, Gokalp G, Erdogan C, Parlak M (2006) The contribution of diffusion-weighted MR imaging to distinguishing typical from atypical meningiomas. Neuroradiology 48(8):513–520
Wibom C, Moren L, Aarhus M, Knappskog PM, Lund-Johansen M, Antti H, Bergenheim AT (2009) Proteomic profiles differ between bone invasive and noninvasive benign meningiomas of fibrous and meningothelial subtype. J Neurooncol 94(3):321–331
Cho YD, Choi GH, Lee SP, Kim JK (2003) (1)H-MRS metabolic patterns for distinguishing between meningiomas and other brain tumours. J Magn Reson Imaging 21:663–672
Filippi CG, Edgar MA, Ulug AM, Prowda JC, Heier LA, Zimmerman RD (2001) Appearance of meningiomas on diffusion-weighted images: correlating diffusion constants with histopathologic findings. Am J Neuroradiol 22:65–72
Majos C, Alonso J, Aguilera C, Serrallonga M, Coll S, Acebes JJ, Arús C, Gili J (2003) Utility of proton MR spectroscopy in the diagnosis of radiologically atypical intracranial meningiomas. Neuroradiology 45:129–136
Shino A, Nakasu S, Matsuda M, Handa J, Morikawa S, Inubushi T (1999) Noninvasive evaluation of the malignant potential of intracranial meningiomas performed using proton magnetic resonance spectroscopy. J Neurosurg 91:928–934
Ferrante L, Acqui M, Artico M, Mastronardi L, Fortuna A (1989) Paediatric intracranial meningiomas. Br J Neurosurg 3:189–196
Artico M, Ferrante L, Cervoni L, Colonnese C, Fortuna A (1995) Pediatric cystic meningioma: a report of three cases. Childs Nerv Syst 11:137–140
Ferrante L, Acqui M, Artico M, Mastronardi L, Rocchi G, Fortuna A (1989) Cerebral meningiomas in children. Childs Nerv Syst 5:83–86
Symons P, Tobias V, Pereira J, Vonau M (2001) Brain-invasive meningioma in a 16-month-old boy. Pathology 33:252–256
Zankl H, Zang KD (1980) Correlations between clinical and cytogenetical data in 180 human meningiomas. Cancer Genet Cytogenet 1:351–356
Zang KD (1982) Cytological and cytogenetical studies on human meningioma. Cancer Genet Cytogenet 2:166–168
Hankins GR, Sasaki T, Lieu AS, Saulle D, Karimi K, Li JZ, Helm GA (2008) Identification of the deleted in liver cancer 1 gene, DLC1, as a candidate meningioma tumor suppressor. Neurosurgery 63(4):771–780
Perry A, Louis DN, Scheithauer BW, Budka H, von Deimling A (2007) Meningeal tumours. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumours of the central nervous system. IARC, Lyon, pp 164–172
Hanft S, Canoll P, Bruce JN (2010) A review of malignant meningiomas: diagnosis, characteristics, and treatment. J Neurooncol 99:433–443
Surace EI, Lusis E, Murakami Y, Scheithauer BW, Perry A, Gutmann DH (2004) Loss of tumor suppressor in lung cancer-1 (TSLC1) expression in meningioma correlates with increased malignancy grade and reduced patient survival. J Neuropathol Exp Neurol 63(10):1015–1027
Liu Y, Pang JC, Dong S, Mao B, Poon WS, Ng HK (2005) Aberrant CpG island hypermethylation profile is associated with atypical and anaplastic meningiomas. Hum Pathol 36(4):416–425
Wen PY, Quant E, Drappatz J, Beroukhim R, Norden AD (2010) Medical therapies for meningiomas. J Neurooncol 99:365–378
Ragel BT, Jensen RL (2010) Aberrant signaling pathways in meningiomas. J Neurooncol 99:315–324
Biegel JA, Parmiter AH, Sutton LN, Rorke LB, Emanuel BS (1994) Abnormalities of chromosome 22 in pediatric meningiomas. Genes Chrom Cancer 9:81–87
Wullich B, Mayfrank L, Schwechheimer K, Finke J, Schempp W (1990) Chromosome abnormalities in multiple meningiomas of a child. Genes Chrom Cancer 2:166–168
Karnes PS, Tran TN, Cui MY, Raffel C, Gilles FH, Barranger JA, Ying KL (1989) Cytogenetic analysis of 39 pediatric central nervous system tumors. Cancer Genet Cytogenet 59:12–19
Slavc I, MacCollin MM, Dunn M, Jones S, Sutton L, Gusella JF, Biegel JA (1995) Exon scanning for mutations of the NF2 gene in pediatric ependymomas, rhabdoid tumours and meningiomas. Int J Cancer 64:243–247
Begnami MD, Rushing EJ, Santi M, Quezado M (2007) Evaluation of NF2 gene deletion in pediatric meningiomas using chromogenic in situ hybridization. Int J Surg Pathol 15(2):110–115
Zwerdling T, Dothage J (2002) Meningiomas in children and adolescents. J Pediatr Hematol Oncol 24(3):199–204
Gondi V, Tome WA, Mehta MP (2010) Fractionated radiotherapy for intracranial meningiomas. J Neurooncol 99:349–356
NICE (2006) Improving outcomes for people with brain and other CNS tumours. Guidance on Cancer Services
Sheehan JP, Williams BJ, Yen CP (2010) Stereotactic radiosurgery for WHO grade I meningiomas. J Neurooncol 99:407–416
Li X, Zhao J (2009) Intracranial meningiomas of childhood and adolescence: report of 34 cases with follow-up. Childs Nerv Syst 25(11):1411–1417
Rogers L, Gilbert M, Vogelbaum MA (2010) Intracranial meningiomas of atypical (WHO grade II) histology. J Neurooncol 99:393–405
Erdincler P, Lena G, Sarioglu AC, Kuday C, Choux M (1998) Intracranial meningiomas in children: review of 29 cases. Surg Neurol 49:136–141
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kotecha, R.S., Junckerstorff, R.C., Lee, S. et al. Pediatric meningioma: current approaches and future direction. J Neurooncol 104, 1–10 (2011). https://doi.org/10.1007/s11060-010-0503-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-010-0503-3