
Abstract
Objective
The goal of this study was to determine the epidemiology, clinical presentation, associated factors, pathological features, and treatment outcome of pediatric meningiomas in a single-center institution.
Methods
Clinical data of 15 patients under 18 years of age operated on for meningiomas from January 1994 to December 2010 were reviewed.
Results
The study group included nine males and six females (mean age of 13 years at surgery). The most common symptoms at presentation were headaches in 6 out of 15 (40 %), raised intracranial pressure in 3 out of 15 (20 %), and seizures in 3 out of 15 (20 %). Sole operated tumors were found in 12 out of 15 (80 %), whose location is as follows: parasagittal in 4 out of 12 (33.3 %), 2 in the convexity (16.6 %), 2 at the skull base (16.6 %), and 4 in other sites (33.3 %). Six children presented with radiation-induced (RT) meningiomas and five had evidence of neurofibromatosis type 2 (NF2). Three patients had multiple meningiomas (all of them had NF2). Simpson’s grade I excision was achieved in 12 out of 15 (80 %). On histopathology, 11 out of 15 (73.3 %) were grade I and 4 out of 15 (26.6 %) were grade II (all of them atypical). Five tumors (33.3 %) recurred, four of which had RT or NF2. During the mean follow-up period of 5 years, 12 out of 15 (80 %) had a good outcome (GOS = 5).
Conclusions
Childhood meningiomas are uncommon lesions with a slight male predominance. Absence of large series with long follow-up precludes any definite conclusions on the clinical course and outcome of these tumors. Associated factors (such as RT and NF2), location, and extent of excision appear to be more important than histopathological grade in predicting outcome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Meningiomas are uncommon childhood tumors. Cushing and Eisenhardt’s 1938 monograph meticulously categorized meningiomas, including their presentation, clinical outcome, and surgical strategies, and recognized the occurrence of child and adolescent meningioma separately [7].
Since then, several series have been published. In adults, meningiomas roughly account for 30 % of primary central nervous system (CNS) neoplasms and extensive research has been directed towards these tumors [21]. On the other hand, child and adolescent meningiomas are rare, accounting for 0.4–4.6 % of CNS tumors in this population, and published data regarding their features are scarce [10, 22].
Management approaches have been drawn from such reports and extrapolated from the treatment of adult meningiomas [45], without clear, statistically validated guidelines for the management of child and adolescent meningiomas. Most tumors are sporadic. However, risk factors for the development of meningiomas include history of radiation therapy (RT) or diagnosis of neurofibromatosis type 2 (NF2) [3, 12, 32, 35, 40].
This report is an attempt to evaluate clinical and pathological features and surgical outcome in a single-center series of pediatric patients who underwent surgery for the treatment of meningiomas in the context of published literature.
Clinical material and methods
Patient population
We reviewed the files of 490 children younger than 18 years harboring a brain tumor, over a 16-year period, who underwent surgery in our institution from January 1994 to December 2010. Fifteen cases of meningiomas were identified and enrolled in the study.
The variables analyzed included age, sex, clinical presentation, radiological features, extent of resection, associated factors (NF2/RT), and histopathology. Extent of resection was analyzed based on the Simpson grading system [43] and the Glasgow Outcome Scale (GOS) was used to analyze outcome.
They had all been preoperatively evaluated by structural brain imaging [axial computerized tomography and/or magnetic resonance (MR) imaging]. Shortly after surgical excision, all patients were put on regular follow-up.
This study was approved by the Research Ethics Committee of the University Hospital of Ribeirão Preto Medical School—USP (Proc. 6591/2007).
Results
Clinical and surgical data
Fifteen patients with meningiomas were identified in a 16-year period, yielding a 3 % occurrence of meningioma among all newly diagnosed brain tumors in children. In the same period, 322 patients with meningiomas had undergone surgery in our institution. Thus, childhood meningiomas accounted for 4.6 % of total meningiomas.
Mean age at diagnosis was 12 years (range, 4–18 years), while mean age at surgery was 13 years (range, 4–18 years). Patients operated on included nine males and six females with a male/female ratio of 1.5:1. The mean length of symptoms/signs ranged from 2 months to 3 years (mean, 12 months). Six patients (40 %) presented with radiation-induced meningiomas and five (33.3 %) had evidence of associated NF2. Patient’s age, gender, tumor location, associated factors, histopathological aspects, and outcome are summarized in Table 1.
The most common presenting symptoms were headaches (40 %), followed by raised intracranial pressure (20 %) and seizures (20 %).
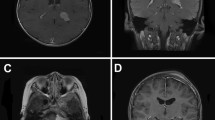
Single tumors were found in 12 out of 15 (80 %), whose location is as follows: parasagittal in 4 out of 12 (33.3 %), convexity in 2 out of 12 (16.6 %), skull base in 2 out of 12 (16.6 %), sphenoidal in 1 (8.3 %), falcotentorial in 1 (8.3 %), intraventricular in 1 (8.3 %), and dorsal spine (D4–D5) in 1 case (8.3 %). Three patients (20 %) had multiple meningiomas, all of which had NF2. Radiological features were similar to adult meningiomas, and cystic changes were observed in only one patient (case 9) (Fig. 1).

Examples of scans of pediatric meningiomas. a Axial enhanced T 1-weighted MR showing a large frontoparietal cystic meningioma in a 5-year-old girl (case 9); b sagittal T 2-weighted MR showing an intradural D4–D5 meningioma (arrow) displacing the spinal cord in a 6-year-old boy with NF2 (case 5); c axial enhanced T 1-weighted MR showing an intraventricular meningioma in an 11-year-old girl (case 10); d parasagittal enhanced T 1-weighted MR showing a jugular foramen meningioma (arrow) in a 12-year-old boy with NF2 (case 7)
A total of 20 meningiomas were operated on in 15 patients. Simpson’s grade I excision could be achieved in 12 patients (80 %), whereas grade II was achieved in 2 patients, and grade III was achieved in 1 patient (case 7, jugular foramen meningioma). Histopathological analysis revealed 11 out of 15 cases (73.3 %) of WHO grade I tumors: 5 transitional, 4 fibrous, and 2 meningotheliomatous. Four (26.6 %) were grade II (all of them atypical). Dura mater and adjacent cortex infiltration were observed in two patients (cases 11 and 14, respectively). There were no postoperative deaths or significant complications in this series.
Out of the five tumors (33.3 %) which recurred, three had previously undergone Simpson’s grade I excision. Two of them presented as radiation-induced meningiomas (cases 1 and 8), and one had NF2 (case 11) (Fig. 2). The other two patients with recurrent lesions underwent a grade II excision (cases 5 and 9). Four of the recurrent meningiomas were reoperated on, and one patient (case 11) was treated with radiosurgery for a small recurrence in the orbit.

Case 8: follow-up MR scans obtained in a patient with a radiation-induced meningioma that developed 11 years after craniospinal RT for medulloblastoma. a Axial enhanced T 1-weighted MR scan showing a large frontoparietal parasagittal meningioma; b post-resection status showing no residual lesion (Simpson grade I); c follow-up axial enhanced T 1-weighted MR scan 12 months post-surgery showing recurrence of the tumor (arrow)
The mean follow-up period was 5 years (range, 1–12 years). At the time of last follow-up assessment, 12 patients had a good outcome (GOS = 5) and 3 had a poor outcome (GOS = 3). Of the three patients with poor outcome, two had previous RT for other lesions at a younger age (cases 1 and 15) and one child with NF2 was severely disabled due to the multiple intracranial lesions (case 4).
Radiation-induced meningioma
Six patients received high-dose (HD) RT (treatment was >20 Gy) for various diseases. Patient demographics and treatment options for the six radiation-induced meningiomas are listed in Table 2. Patients’ age when the radiation-induced meningiomas were diagnosed ranged from 13 to 18 years (mean, 16.1 years). The latency or induction period ranged from 10 to 13 years (mean, 11 years).
Cytogenetic studies
Cytogenetic data were available for three patients. While case 9 showed a normal 46, XX karyotype, two radiation-induced meningiomas showed complex aberrations. Case 9 showed a composite karyotype with aberrations involving chromosomes 1, 6, and 12 (previously published by our group) [5]. Case 15, on the other hand, exhibited a clonal aberration involving chromosomes 1 and 3, with a karyotype denoted as 46, XX, t(1;3)(p22;q12), del(1)(p?)[8]/46, XX[12].
Discussion
Meningiomas are uncommon neoplasms in the pediatric age group and differ in various clinical and biological aspects from meningiomas in the adult population [4, 31]. It has been reported that their frequency was <5 % of all pediatric brain tumors [18]. In our series, the incidence of pediatric meningiomas was 3 %.
Different from adults in whom meningiomas are twice more common in women than in men, some previous studies showed no gender predilection in the pediatric group [3, 46]. However, the present series showed a male preponderance of this tumor and similar results were documented by other authors [11, 31]. This greater occurrence of meningiomas in males could be related to the absence of the effect of sex hormones on steroid receptors in meningioma cells, for children have lower hormonal serum levels [9, 31].
Childhood meningiomas are characteristically known to have nonspecific symptoms and diagnosis is often difficult. Common clinical manifestations of pediatric meningiomas include signs of increased intracranial pressure, focal neurological deficits, seizures, and other signs based on their location [32].
Kotecha et al. [22] published a meta-analysis and observed that headaches, seizures, and vomiting were the most common symptoms, occurring in 45.6, 32.9, and 29.7 % of patients, respectively. In our series, headache was the most common symptom (40 % of cases). Signs and symptoms related to increased intracranial pressure were present in only 20 % of our cases. The incidence of meningiomas increases with age and a larger number of cases is reported in the second decade of life compared to the first one [21]. Only 3 out of 15 children in our series were below the age of 10 years. The mean age at onset in our series was 12 months.
We conducted a literature review of 668 pediatric meningiomas (with an upper age limit of 20 years) published in different series, ranging from 6 to 87 cases [1, 3, 4, 8, 9, 11, 13, 15, 17, 18, 20, 27–30, 32, 35, 38–40, 44, 46, 47, 50, 51]. Table 3 summarizes the cases of pediatric meningioma.
Tumors were most commonly intracranial, making up to 89.8 % of the cases; the remaining cases were intraspinal in 5.8 %, intraorbital in 1.9 %, and located elsewhere in 2.3 %. The cerebral convexity was the most frequent location, followed by intraventricular, falcine and parasagittal, and anterior and middle cranial fossa.
In the present series, the most common location was parasagittal (40 %). We observed only one case with a tumor in the dorsal spine. However, pediatric meningiomas are known to occur in uncommon sites like the skull base and posterior fossa [11, 32, 36]. There is a greater incidence of intraventricular meningiomas in children as reported in the literature (11.3 % [6] vs. 0.5–2 % [26]). We observed an intraventricular lesion and one skull base (jugular foramen) meningioma.
Multifocal meningiomas at presentation occurred in three patients (20 %). All the three patients had NF2. A meta-analysis confirmed a significantly higher risk of multiple meningiomas in patients with NF2 [22].
The pattern of histopathological variants seen in our series differed significantly from previous data available in the literature [8, 11, 28]. Childhood meningiomas are known to have a high incidence of atypical histopathology, especially the clear cell and the papillary variants. We did not have any clear cell or papillary variants even though four patients (26.6 %) had atypical meningiomas. All of them presented with associated factors: three cases of radiation-induced meningiomas and one patient with NF2. Overall, previous reports have showed a high incidence of atypical or anaplastic histopathology [4, 14, 31, 32].
Perry et al. [36] noted a high frequency of brain invasion in pediatric meningiomas and reported them to be phenotypically and genotypically aggressive when compared with adults. In our series, two patients (13.3 %) presented with dura mater and adjacent cortical invasion.
The association between NF2 and meningiomas is well known, and they may share common mechanisms of pathogenesis. The possibility of NF2 should be kept in mind when treating a child with a meningioma and approximately 25–40 % of children with meningiomas have NF2 mutations [36, 40]. The large size of some pediatric meningiomas (as mentioned in several series), especially those related to NF lineages, suggests that tumor growth is rapid in children [3].
Loss of NF2 gene expression (which is located on 22q12) occurs in almost all NF2-associated meningiomas and 40–60 % of sporadic meningiomas [35]. In a review of the literature, NF was present in 89 cases (13.3 %), and NF2 was the most common, occurring in 57 out of 89 patients (64 %). Two large studies of pediatric meningiomas yielded incidences of 41 and 45.4 %, respectively, for NF [11, 36], with spinal and optic nerve sheath meningiomas being the most common ones. Five children (33.3 %) in our series had evidence of associated NF2. Patients with NF2 have a significantly worse prognosis and a substantial change in overall survival, compared with patients without neurofibromatosis [21]. Likewise, patients with NF2 are more prone to develop multiple meningiomas. Since the risks of any treatment need to be balanced with the natural history of the disease, these tumors are often not aggressively resected. Both these factors could account for the decline in overall survival in NF2 patients after 10 years and worse prognosis compared with patients without neurofibromatosis [21].
The causal relationship between radiation and pediatric meningioma is well established. The high sensitivity of the arachnoid membranes to irradiation in children increases vulnerability to oncogenic stimulation. Ionizing radiation causes mutations in the genome, either directly or indirectly through the formation of free radicals [22].
Radiation-induced meningiomas typically present at an earlier age, arise within the prior irradiation field, and are more likely to be multifocal and exhibit higher degrees of atypia and mitosis [16]. It has also been noted that radiation-induced meningiomas differ from nonradiation-induced meningiomas in several key aspects. They tend to behave more aggressively, possess atypical histological features, and display more rapid growth and higher rates of multiplicity and recurrence than meningiomas not induced by RT [2, 32, 48].
There is also some suggestion of a dose-related effect, with higher levels of radiation exposure being associated with shorter latency periods for development of meningiomas [19, 49]. Harrison et al. [19] classified radiation-induced meningiomas into three groups based on the amount of radiation administered, as follows: (1) low dose (LD) (<10 Gy); (2) moderate dose (10–20 Gy); and (3) high dose (>20 Gy).
However, it has been reported that meningiomas occur more frequently after LD radiation. Al-Mefty et al. [2] reported that the interval between radiotherapy and detection of the meningioma was inversely related to dose. The difference in latency between tumors treated with HD and LD RT indicates that the more severe chromosome injury caused by higher radiation doses elicits more rapid loss of cellular control mechanisms and earlier expression of the neoplastic phenotype [4, 41]. Less than 100 radiation-induced meningioma cases in pediatric patients have been reported in the medical literature since 1953 [4, 37]. Six patients (40 %) in our series received HD RT and developed meningiomas at a later stage. The mean latency or induction period was 11 years.
It is widely accepted that tumor progression results from chromosome instability and genetic variability within the tumor cell population, which allows for clonal expansion of more aggressive tumor phenotypes [34]. Primary cytogenetic changes in pediatric meningiomas are similar to those found in the adult counterparts, with loss of chromosome 22 (in patients with NF2), followed by loss of 1p, being the most frequent changes [42]. Also partial or complete loss of the short arm of chromosome 1 seems to be strictly correlated with the grading of meningiomas [33]. Interestingly, both radiation-induced tumors analyzed by our group showed aberrations in the short arm of chromosome 1. Although radiation-induced meningiomas often have complex structural and numerical chromosomal abnormalities consistent with DNA damage induced by irradiation [25], it has been suggested that the loss of 1p as a consequence of irradiation may be more important in the development of meningiomas than other chromosomal lesions [42]. Moreover, the data showed herein demonstrate a positive correlation between chromosome instability and proliferation, as proved by the increased Ki-67 index observed in both cases with high chromosome instability (case 8 with 20 % and case 15 with 5–10 %).
The goal of treatment for meningiomas is total resection with wide dural clearance and re-excision should be considered in the event of initial subtotal resection. Resection of the dural origin/attachment is recommended, since there is a higher recurrence risk if the dural attachment is left behind [22, 40].
The surgical treatment of these tumors in children poses a major challenge in neurosurgical practice. The difficult location of these tumors, larger size at presentation, relatively less blood volume in children, and the risks of lengthy operations such as hypothermia and massive blood transfusion all contribute to problems in surgical management [32].
Benign meningiomas are associated with high cure rates, provided that the entirety of the tumor is excised. However, there is always a considerable risk of recurrence even after gross total resection with excision of the surrounding dura and involved bone is achieved, suggesting that biological heterogeneity exists among benign lesions [1, 14]. In the present series, gross total resection (Sympson’s grade I) was achieved in 12 patients (80 %).
Due to the lack of evidence in children and adults, guidelines for radiotherapy are limited and use varies significantly between institutions. In children, the use of radiotherapy must be balanced with the risk of significant late sequelae. This is particularly important in infants and young children whose CNS is more vulnerable to the effects of radiotherapy [37, 49].
The clinical evolution of meningiomas in children is not reliably predictable and remains an issue. In the medical literature, recurrence was observed in 20.3 %, ranging in different series from 0 to 40.9 %. In our series, recurrence occurred in five patients (33.3 %). Four of them had associated factors.
The prognosis of pediatric meningiomas has improved over the years following advances in surgical techniques and supportive care [22]. The largest case series of 87 pediatric meningiomas reported a mortality rate of 10.6 % and a recurrence rate of 19.4 % [40]. There are 81 deaths reported in the literature (12.1 %), ranging from 0 to 50 %. Our patients had a more favorable outcome, and during the mean follow-up period of 5 years, there were no deaths and the majority of patients (80 %) had a good outcome.
Conclusions
Intracranial meningiomas are uncommon in the pediatric population. They have a male predominance and show higher rates of atypical features when compared with adult meningiomas. Total resection is associated with a better prognosis and should always be attempted. RT should be reserved to selected cases.
Location, associated factors (RT/NF2), and extent of excision appear to be more important than histology in predicting outcome. Further studies are required to establish the role of proliferative indexes and biological markers in pediatric meningiomas.
References
Alexiou GA, Mpairamidis E, Psarros A, Sfakianos G, Prodromou N (2008) Intracranial meningiomas in children: report of 8 cases. Pediatr Neurosurg 44:373–75
Al-Mefty O, Topsakal C, Pravdenkova S, Sawyer JR, Harrison MJ (2004) Radiation-induced meningiomas: clinical, pathological, cytokinetic, and cytogenetic characteristics. J Neurosurg 100:1002–13
Amirjamshidi A, Mehrazin M, Abbassioun K (2000) Meningiomas of the central nervous system occurring below the age of 17: report of 24 cases not associated with neurofibromatosis and review of literature. Childs Nerv Syst 16:406–16
Arivazhagan A, Devi BI, Kolluri SV, Abraham RG, Sampath S, Chandramouli BA (2008) Pediatric intracranial meningiomas—do they differ from their counterparts in adults? Pediatr Neurosurg 44:43–48
Brassesco MS, Valera ET, Neder L, Castro-Gamero AM, de Oliveira FM, Santos AC, Scrideli CA, Oliveira RS, Machado HR, Tone LG (2009) Childhood radiation-associated atypical meningioma with novel complex rearrangements involving chromosomes 1 and 12. Neuropathology 29:585–90
Choux M, Lena G, Genitoro L, Azambuja N, Cunha C, De-chambenoit G (1991) Meningiomas in children. In: Schmidek HH (ed) Meningiomas and their surgical management. WB Saunders, Philadelphia, pp 93–102
Cushing H, Eisenhardt L (1938) Serial enumeration of meningiomas. In: Thomas CC, (ed) Meningiomas: their classification, regional behaviour, life history, and surgical end results. Classics of Neurology & Neurosurgery Library, Springfield, IL pp 56–73
Davidson GS, Hope JK (1989) Meningeal tumors of childhood. Cancer 63:1205–10
Deen HG, Scheithauer BW, Ebersold MJ (1982) Clinical and pathological study of meningiomas of the first two decades of life. J Neurosurg 56:317–322
Di Rocco C, Di Rienzo A (1999) Meningiomas in childhood. Crit Rev Neurosurg 9:180–88
Erdinçler P, Lena G, Sarioglu AC, Kuday C, Choux M (1998) Intracranial meningiomas in children: review of 29 cases. Surg Neurol 49:136–140, discussion 140–131
Evans DGR, Watson C, King A, Wallace AJ, Baser ME (2005) Multiple meningiomas: differential involvement of the NF2 gene in children and adults. J Med Genet 42:45–48
Ferrante L, Acqui M, Artico M, Mastronardi L, Rocchi G, Fortuna A (1989) Cerebral meningiomas in children. Childs Nerv Syst 5:83–86
Jaiswal S, Vij M, Mehrotra A, Jaiswal AK, Srivastava AK, Behari S (2011) A clinicopathological and neuroradiological study of paediatric meningioma from a single centre. J Clin Neurosci 18:1084–9
Gao X, Zhang R, Mao Y, Wang Y (2009) Childhood and juvenile meningiomas. Childs Nerv Syst 25:1571–80
Galloway TJ, Indelicato DJ, Amdur RJ, Swanson EL, Morris CG, Marcus RB (2011) Favorable outcomes of pediatric patients treated with radiotherapy to the central nervous system who develop radiation-induced meningiomas. Int J Radiat Oncol Biol Phys 79:117–20
Germano IM, Edwards M, Davis RL, Schiffer DJ (1994) Intracranial meningiomas of the first two decades of life. J Neurosurg 80:447–453
Greene S, Nair N, Ojemann JG, Ellenbogen RG, Avellino AM (2008) Meningiomas in children. Pediatr Neurosurg 44:9–13
Harrison MJ, Wolfe DE, Lau TS, Mitnick RJ, Sachdev VP (1991) Radiation-induced meningiomas: experience at the Mount Sinai Hospital and review of the literature. J Neurosurg 75:564–74
Im SH, Wang KC, Kim SK, Oh CW, Kim DG, Hong SK, Kim NR, Chi JG, Cho BK (2001) Childhood meningioma: unusual location, atypical radiological findings, and favorable treatment outcome. Childs Nerv Syst 17:656–62
Kotecha RS, Junckerstorff RC, Lee S, Cole CH, Gottardo NG (2011) Pediatric meningioma: current approaches and future direction. J Neurooncol 104:1–10
Kotecha RS, Pascoe EM, Rushing EJ, Rorke-Adams LB, Zwerdling T, Gao X, Li X, Greene S, Amirjamshidi A, Kim SK, Lima MA, Hung PC, Lakhdar F, Mehta N, Liu Y, Devi BI, Sudhir BJ, Lund-Johansen M, Gjerris F, Cole CH, Gottardo NG (2011) Meningiomas in children and adolescents: a meta-analysis of individual patient data. Lancet Oncol 12:1229–39
Lakhdar F, Arkha Y, El Ouahabi A, Melhaoui A, Rifi L, Derraz S, El Khamlichi A (2010) Intracranial meningioma in children: different from adult forms? A series of 21 cases. Neurochirurgie 56:309–14
Li X, Zhao J (2009) Intracranial meningiomas of childhood and adolescence: report of 34 cases with follow-up. Childs Nerv Syst 25:1411–17
Lillehei KO, Donson AM, Kleinschmidt-DeMasters BK (2008) Radiation-induced meningiomas: clinical, cytogenetic and microarray features. Acta Neuropathol 116:289–301
Liu M, Wei Y, Liu Y, Zhu S, Li X (2006) Intraventricular meningiomas: a report of 25 cases. Neurosurg Rev 29:36–40
Liu Y, Li F, Zhu S, Liu M, Wu C (2008) Clinical features and treatment of meningiomas in children: report of 12 cases and literature review. Pediatr Neurosurg 44:112–17
Lund-Johansen M, Scheie D, Muller T, Lundar T, Helseth E (2001) Neurosurgical treatment of meningiomas in children and young adults. Childs Nerv Syst 17:719–23
Mallucci CL, Parkes SE, Barber P, Powell J, Stevens MCG, Walsh AR, Hockley AD (1996) Paediatric meningeal tumours. Childs Nerv Syst 12:582–89
Maranhão-Filho P, Campos JC, Lima MA (2008) Intracranial meningiomas in children: ten-year experience. Pediatr Neurol 39:415–17
Mehta N, Bhagwati S, Parulekar G (2009) Meningiomas in children: a study of 18 cases. J Pediatr Neurosci 4:61–65
Menon G, Nair S, Sudhir J, Rao BR, Mathew A, Bahuleyan B (2009) Childhood and adolescent meningiomas: a report of 38 cases and review of literature. Acta Neurochir 151:239–44
Müller P, Henn W, Niedermayer I, Ketter R, Feiden W, Steudel WI, Zang KD, Steilen-Gimbel H (1999) Deletion of chromosome 1p and loss of expression of alkaline phosphatase indicate progression of meningiomas. Clin Cancer Res 5:3569–77
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194:23–8
Perry A, Dehner LP (2003) Meningeal tumours of childhood and infancy. An update and literature review. Brain Pathol 13:386–408
Perry A, Giannini C, Raghavan R, Scheithauer BW, Banerjee R, Margraf L, Bowers DC, Lytle RA, Newsham IF, Gutmann DH (2001) Aggressive phenotypic and genotypic features in paediatric and NF2-associated meningiomas: a clinico-pathologic study of 53 cases. J Neuropathol Exp Neurol 60:994–1003
Pettorini BL, Park YS, Caldarelli M, Massimi L, Tamburrini G, Di Rocco C (2008) Radiation-induced brain tumours after central nervous system irradiation in childhood: a review. Childs Nerv Syst 24:793–805
Raaschou-Nielsen O, Sørensen M, Carstensen H, Jensen T, Bernhardtsen T, Gjerris F, Schmiegelow K (2006) Increasing incidence of childhood tumours of the central nervous system in Denmark, 1980–1996. Br J Cancer 95:416–22
Rochat P, Johannesen H, Gjerris F (2004) Long term follow up of children with meningiomas in Denmark 1935–1984. J Neurosurg 100(Pediatrics 2 Suppl):179–182
Rushing EJ, Olsen C, Mena H, Rueda ME, Lee YS, Keating RF, Packer RJ, Santi M (2005) Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg 103(Pediatrics 6 Suppl):489–95
Salvati M, Cervoni L, Puzzilli F, Bristot R, Delfini R, Gagliardi FM (1997) High-dose radiation-induced meningiomas. Surg Neurol 47:435–42
Shoshan Y, Chernova O, Juen SS, Somerville RP, Israel Z, Barnett GH, Cowell JK (2000) Radiation-induced meningioma: a distinct molecular genetic pattern? J Neuropathol Exp Neurol 59:614–20
Simpson D (1957) The Recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiat 20:22–39
Teixidor P, Guillén A, Cruz O, Costa JM (2008) Intracranial meningiomas in children: report of 10 cases. Neurocirugia 19:434–39
Traunecker H, Mallucci C, Grundy R, Pizer B, Saran F (2008) Children’s Cancer and Leukaemia Group (CCLG): guidelines for the management of intracranial meningioma in children and young people. Br J Neurosurg 22:13–25
Tufan K, Dogulu F, Kurt G, Emmez H, Ceviker N, Baykaner MK (2005) Intracranial meningiomas of childhood and adolescence. Pediatr Neurosurg 41:1–7
Turgut M, Ozcan O, Bertan V (1997) Meningiomas in childhood and adolescence: a report of 13 cases and review of literature. Br J Neurosurg 11:501–507
Umansky F, Shoshan Y, Rosenthal G, Fraifeld S, Spektor S (2008) Radiation-induced meningioma. Neurosurg Focus 24:E7
Vinchon M, Leblond P, Caron S, Delestret I, Baroncini M, Coche B (2011) Radiation-induced tumors in children irradiated for brain tumor: a longitudinal study. Childs Nerv Syst 27:445–53
Yoon HK, Kim SS, Kim IO, Na DG, Byun HS, Shin HJ, Han BK (1999) MRI of primary meningeal tumours in children. Neuroradiology 41:512–6
Zwerdling T, Dothage J (2002) Meningiomas in children and adolescents. J Pediatr Hematol/Oncol 24:199–204
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Santos, M.V., Furlanetti, L., Valera, E.T. et al. Pediatric meningiomas: a single-center experience with 15 consecutive cases and review of the literature. Childs Nerv Syst 28, 1887–1896 (2012). https://doi.org/10.1007/s00381-012-1823-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-012-1823-8