
Abstract
Maintaining genome stability is essential to an organism’s health and survival. Breakdown of the mechanisms protecting the genome and the resulting genome instability are an important aspect of the aging process and have been linked to diseases such as cancer. Thus, a large network of interconnected pathways is responsible for ensuring genome integrity in the face of the continuous challenges that induce DNA damage. While these pathways are diverse, epigenetic mechanisms play a central role in many of them. DNA modifications, histone variants and modifications, chromatin structure, and non-coding RNAs all carry out a variety of functions to ensure that genome stability is maintained. Epigenetic mechanisms ensure the functions of centromeres and telomeres that are essential for genome stability. Epigenetic mechanisms also protect the genome from the invasion by transposable elements and contribute to various DNA repair pathways. In this review, we highlight the integral role of epigenetic mechanisms in the maintenance of genome stability and draw attention to issues in need of further study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ensuring the integrity of the genome while maintaining the flexibility to allow for adaptive change through mutation is essential for the long-term survival of a species. Genomes are assaulted continuously by various forces impacting their integrity, including both internal and external factors (Tubbs and Nussenzweig 2017). Examples of external forces include DNA damaging ionizing or UV radiation as well as various chemicals such as oxidizing agents, DNA intercalators, and base analogs (Chatterjee and Walker 2017; Mehta and Haber 2014). Viruses that can integrate into host genomes, thus potentially disrupting vital gene functions, are another external factor impacting genome stability (Weitzman and Fradet-Turcotte 2018). Internal forces affecting genome integrity include, among others, the error rate of the DNA replication machinery as well as the efficiency of DNA repair pathways, reactive oxidant byproducts of metabolism, and active transposable elements (TEs) (Aguilera and Garcia-Muse 2013). Thus, to keep their genome intact and ensure genome stability, organisms have to manage these diverse challenges while tolerating the low level of mutation that is the basis of evolutionary change.
The importance of genome stability is illustrated further by the consequences of its breakdown. Genome instability occurs when the genome accumulates mutations at an increased rate (Aguilera and Garcia-Muse 2013). It is a hallmark of many cancerous tissues, which tend to exhibit numerous genetic alterations compared to the genomes of matched non-cancerous tissues (Tubbs and Nussenzweig 2017). The genetic alterations observed in cancers are diverse and can include single-base mutations, copy number changes, chromosomal rearrangements, and abnormal chromosome numbers (Sansregret et al. 2018). Genome instability also has been associated with the aging process, as mutation rates tend to increase in senescent cells and with increased organismal age (Vijg and Suh 2013). In addition, rates of TE transposition have been reported to increase with aging, contributing significantly to genome instability (Li et al. 2013; Pal and Tyler 2016). These examples demonstrate that genome stability is of vital importance to ensure organismal health, in addition to being key for the long-term survival of a species.
Given the variety of factors able to damage DNA and destabilize genomes, maintaining genome integrity is a complex problem, and numerous intersecting pathways contribute to genome stability. In humans, one example of a pathway protecting the genome from external factors is the production of melanin in the skin (Brenner and Hearing 2008). Melanin protects the genome of skin cells from UV radiation-induced damage, thus preventing mutations and leading to a lower skin cancer rate in individuals with higher levels of melanin (Brenner and Hearing 2008). Another example of pathways contributing to genome stability are the DNA repair pathways that repair DNA double-strand breaks, including the non-homologous end joining (NHEJ) pathway and the homologous recombination (HR) pathway (Chang et al. 2017; Wright et al. 2018). DNA double-strand breaks can be caused by both internal and external factors, but the NHEJ repair pathway, for example, is used for their repair irrespective of the cause of the DNA double-strand break. The use of the NHEJ repair pathway is triggered when a DNA double-strand break is detected and the DNA end is suitable for repair via this pathway, which uses ligation to repair the DNA break without referring to a homologous DNA template (Scully et al. 2019). These examples illustrate that there are diverse mechanisms contributing to genome integrity, which collectively ensure genome stability.
Several of the mechanism and pathways ensuring genome stability are linked to epigenetics. Epigenetics encompasses a variety of phenomena and molecular systems that are connected by the fact that they involve heritable changes in phenotypes or gene expression that are independent of changes in DNA sequence (Felsenfeld 2014). Thus, epigenetics includes the study of DNA methylation, histone modifications, chromatin structure, and non-coding RNAs, both large and small (Felsenfeld 2014) Interestingly, many of these molecular mediators of epigenetic inheritance have been linked to genome stability, and they contribute to varying degrees to the maintenance of genome integrity (Felsenfeld 2014; Fischer and Riddle 2018). In contrast, genome instability often is associated with the breakdown of epigenetic mechanisms, an observation which further supports the link between epigenetics and genome stability. In this article, we review how epigenetic mechanisms impact genome stability (Fig. 1). We survey the role of epigenetic mechanisms in ensuring centromere and telomere function, their impacts on TE activity, and their interaction with DNA repair pathways. We discuss histone variants and modifications, DNA methylation, and non-coding RNAs and demonstrate the importance of these epigenetic systems in ensuring genome stability. Finally, we provide an assessment of the current gaps in knowledge and the opportunities to address them.

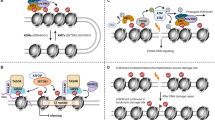
Complex interactions between epigenetics and genome stability. Genome stability is of central importance to organismal survival (middle). Here, we highlight four major facets of genome stability (telomeres, centromeres, TE regulation, and DNA repair; purple) and the epigenetic mechanisms that contribute to these pathways (brown)
Epigenetic mediators are essential for centromere function and genome stability
One strong link between epigenetics and genome stability is provided by centromeres, which have an essential role in maintaining genome stability and are specified by epigenetic mechanisms. Centromeres are the portions of eukaryotic chromosomes where the kinetochore assembles and microtubules attach during meiosis and mitosis (McKinley and Cheeseman 2016). Thus, centromeres ensure proper segregation of chromosomes to daughter cells. Dysfunction of centromeres has severe consequences: it can lead to chromosome breakage and fusion, as well as chromosome loss and aneuploidy (Barra and Fachinetti 2018). In severe cases, cytokinesis failure due to lagging chromosomes can lead to polyploidy, which has the potential to further destabilize the genome (Lens and Medema 2019). Another consequence of centromere dysfunction and lagging chromosomes can be the formation of micronuclei, which make the enclosed DNA susceptible to increased rates of DNA damage, replication errors, and again, further enhancement in genome instability (Chunduri and Storchova 2019). Ultimately, centromere dysfunction leads to genome instability by several mechanisms and, if the appropriate cell cycle checkpoints are triggered, cell death [for an example, see (Caneus et al. 2018)]. Thus, centromeres perform vital functions in ensuring genome stability.
Despite their importance for cell viability and genome stability, centromere organization varies widely among species. Based on their centromere organization, chromosomes can be classified as monocentric, which includes both point centromeres and regional centromeres, or as holocentric (McKinley and Cheeseman 2016; Steiner and Henikoff 2015). Monocentric chromosomes have one centromere, which coincides with the primary constriction visible in a mitotic chromosome; examples of species with monocentric chromosomes include humans as well as several model species such as Drosophila melanogaster, Arabidopsis thaliana, Schizosaccharomyces pombe, and Saccharomyces cerevisiae (point centromeres). In contrast, holocentromeres are not restricted to one region of the chromosome, and there is no constriction visible in the mitotic chromosome (Steiner and Henikoff 2015). Instead of the microtubules attaching at just one specific region of the chromosome, they attach at sites along the entire length of the chromosome (Steiner and Henikoff 2015). Examples of species with holocentromeres include the model organism Caenorhabditis elegans and a variety of plant species (Cuacos et al. 2015). These examples illustrate that there is considerable variation among species in how centromeres are organized.
Despite the essential function of centromeres in the regulation of chromosome segregation during cell division, centromere location is not encoded in the DNA sequence (the point centromeres of S. cerevisiae might be considered an exception to this rule) (McKinley and Cheeseman 2016). This fact is best illustrated by the study of neocentromeres, which are centromeres that form at a new location on the chromosome that has not acted previously as a centromere (Fukagawa and Earnshaw 2014b; Scott and Sullivan 2014). Many regional centromeres, such as the ones found in humans, are associated with specific repeated sequences. In humans, for example, centromeres are formed typically over regions containing alpha-satellite repeats. Similarly, regional centromeres in the fruit fly model Drosophila melanogaster contain various AT-rich repeats such as the 359 bp repeat, and centromeres in the plant model system Arabidopsis thaliana are composed of arrays of a 180-bp repeat (Plohl et al. 2014). When centromere location shifts along the chromosome leading to the formation of a neocentromere, the sequences underlying the neocentromeres are diverse, and the repeats typically associated with the centromere can still be detected at its original location (Amor et al. 2004; Schneider et al. 2016). Neocentromeres have been detected in humans on various chromosomes (Fukagawa and Earnshaw 2014a; Scott and Sullivan 2014; Voullaire et al. 1993), and they have been experimentally induced in several species including Drosophila melanogaster (Maggert and Karpen 2001) and chicken (Shang et al. 2013). Thus, neocentromere studies support the conclusion that DNA sequence does not specify centromere location, but that the centromere is specified instead epigenetically.
Research over several decades has led to a model which suggests that the centromere location is determined by the presence of the centromere-specific histone variant CenH3, also known as CENP-A (Palmer et al. 1991; Scott and Sullivan 2014). In addition to the core histones, H2A, H2B, H3, and H4, which typically make up nucleosomes, variants derived from these histone genes exist in many species (Henikoff and Smith 2015; Talbert and Henikoff 2017). Very few variants exist for H2B and H4, whereas H3 has two major variants: CenH3 and H3.3 discussed below. CenH3 is a variant of histone H3 that, under normal conditions, occurs exclusively at centromeres (Gambogi and Black 2019; Palmer et al. 1991). CenH3 is unusual in that, in contrast to the canonical histones, it is undergoing rapid evolutionary change, with sequence changes quickly accumulating, especially in its amino-terminal tail (Malik and Henikoff 2003; Rosin and Mellone 2017). CenH3-containing nucleosomes are found at the functional centromeres of metacentric and holocentric chromosomes, and the S. cerevisiae point centromeres consist of a single CenH3(Cse4)-containing nucleosome (Choy et al. 2012; Furuyama and Biggins 2007; McKinley and Cheeseman 2016). Neocentromeres also contain CenH3 nucleosomes (Henikoff and Furuyama 2012; Warburton et al. 1997), illustrating the importance of this centromeric histone variant, which epigenetically specifies the functional centromere and is thus essential for genome stability.
Interestingly, as more genome sequences have become available, a number of species were identified that lack a recognizable copy of CenH3 in their genome (Drinnenberg et al. 2016). These species include a group of unicellular flagellar eukaryotes related to trypanosomes (Akiyoshi and Gull 2014; Berriman et al. 2005) and also a variety of insect species, including the Lepidoptera (butterflies) and Odonata (dragon flies) (Drinnenberg et al. 2014). In the insect species lacking CenH3, most other kinetochore proteins are conserved (Drinnenberg et al. 2014), while in the trypanosomes, the conventional kinetochore proteins are absent as well, with novel proteins absorbing their functions (Akiyoshi and Gull 2014). As CenH3 typically is required to specify the centromere location, these findings raise the question of how centromeres form in the species lacking CenH3, and how a protein essential for genome stability can be lost over evolutionary time.
Interestingly, a second variant of H3 also contributes to centromere function. H3.3 differs from canonical H3 by only four to five amino acids, and it is typically found in regions of the genome that are actively transcribed (Dion et al. 2007; Franklin and Zweidler 1977; Henikoff and Smith 2015; Schwartz and Ahmad 2005). Remarkably, genetic studies have shown that it is crucial for maintaining genome stability during mammalian development (Bush et al. 2013; Jang et al. 2015). In mice, complete loss of H3.3 leads to embryonic lethality. Loss of H3.3 in embryonic stem cells leads to mitotic defects, including chromosome bridges and lagging chromosomes in anaphase. These problems during mitosis result in abnormal chromosome number, genome instability, and cell death, which are likely the cause of the embryonic lethality. In addition, H3.3 loss also negatively impacts the chromatin structure at the telomeres, centromeres, and pericentric regions, which contributes to the observed genome instability (Jang et al. 2015). Thus, histone variants are heavily involved in regulating the function of the centromere and contribute significantly to genomic stability.
Studies in several species have shown that while the position of the centromere is determined epigenetically by the presence of CenH3 in most species, the strength of a centromere and its ability to ensure faithful chromosome segregation are determined by genetic as well as epigenetic factors. While centromeres are formed at a specific region along the chromosome in regional monocentric chromosomes, the exact position of where CenH3 localizes is variable among individuals, and the CenH3-containing chromatin block can shift. These regional shifts in CenH3 location have been documented, for example, in the genus Equus by Nergadze and colleagues, who find that centromeric epialleles with slightly different localization of the CenH3 chromatin are quite common (Nergadze et al. 2018). In maize, Gent and colleagues find that the exact location of the centromere position can shift between generations (Gent et al. 2017). In humans, centromeric epialleles have been described for chromosome 17, which differ in the composition of the underlying alpha-satellite sequence, leading to variability in centromere function between the two epialleles (Aldrup-MacDonald et al. 2016; Maloney et al. 2012). Specifically, one of the centromeric epialleles, D17Z1, showed decreased CenH3 recruitment and increased levels of aneuploidy for this chromosome (Aldrup-MacDonald et al. 2016). These studies demonstrate that the centromeric epialleles impact centromere function and can differ significantly in their contribution to genome stability.
Another aspect of the epigenetic state of the centromere that impacts its ability to ensure genome stability is the post-translational modification (PTM) of the histones found at the centromere. PTMs of both CenH3 and the canonical histones present at the centromere are important for its function (Bowman and Poirier 2015; Johnson et al. 2004; Loyola et al. 2006; McKittrick et al. 2004; Waterborg 1990). Canonical histone H3 present at the centromere and interspersed with CenH3 nucleosomes (Blower et al. 2002) is methylated at H3K4 (H3K4me2) and lacks H3K9 methylation (me2 and me3), which is found in the pericentromeric regions (Sullivan and Karpen 2004). Using a human artificial chromosome (HAC) system, Molina and colleagues removed H3K4me2 from the centromere and found that segregation errors for this chromosome increased (Molina et al. 2016). If H3K9me2/me3 chromatin spreads from the pericentric regions into CenH3 chromatin, centromere function is perturbed also, leading to chromosome segregation defects and genome instability (Bergmann et al. 2012; Ohzeki et al. 2016). In addition, CenH3 is subject to PTM (Srivastava and Foltz 2018), and some of the PMTs have been linked to centromere function and genome stability. For example, on CENP-A, the human CenH3 homology, serine 18 (S18) hyperphosphorylation leads to increased genome instability (Takada et al. 2017), while CENP-A serine 7 (S7) phosphorylation is not essential for centromere function (Barra et al. 2019). Together, these findings demonstrate that epigenetic marks in the form of histone PMTs at the centromere are essential for centromere function and the maintenance of genome stability.
Non-coding RNAs are another way epigenetic mechanisms contribute to centromere function and genome stability (Talbert and Henikoff 2018). Volpe and colleagues demonstrated that small RNAs derived from the outer centromeric repeats in S. pombe are required for centromere function (Volpe et al. 2003, 2002). Small RNAs since have been shown to be derived from centromeres in other species as well (Cohen et al. 1973; Hall et al. 2002; Maison et al. 2002; Perea-Resa and Blower 2018), and the data suggest that they are important for centromere function and genome stability. In addition to these small RNA classes, there are also centromere-derived RNAs that appear to be an integral part of the centromere (Ling and Yuen 2019; Talbert and Henikoff 2018). These RNAs were observed first in maize in 2004 (Topp et al. 2004) and since have been the focus of detailed studies. In Drosophila, loss of the long non-coding RNAs from the centromeric 359 bp repeat leads to chromosome segregation defects (Rošić et al. 2014), and Ling and colleagues discovered that in S. cerevisiae both over- and underproduction of the long non-coding RNAs from the centromere leads to increased levels of aneuploidy (Ling and Yuen 2019). Together, the data available suggest that non-coding RNAs, both large and small, are required for the optimal function of the centromere to ensure genome stability.
In summary, centromeres are essential for genome stability as they ensure proper chromosome segregation to daughter cells. Centromere position is epigenetically determined by the location of the CenH3 histone variants, and various epigenetic mechanisms including non-coding RNAs, histone modification, and chromatin structure components are essential for normal centromere function. Thus, studies of centromere biology demonstrate unequivocally that epigenetic mechanisms contribute essential functions to genome stability and how disruptions of these mechanisms lead to genome instability.
Telomeres rely on epigenetic mechanisms to ensure genome stability
In addition to centromeres, telomeres are a second type of chromosomal structure that is essential for maintaining genome stability and that employs epigenetic mechanisms for optimal function. Telomeres are structures at the end of linear chromosomes that protect chromosome ends from fusion by recombination and from degradation by nucleases (O'Sullivan and Karlseder 2010). Typically, telomeres exhibit a specialized chromatin structure that is similar to heterochromatin and imparts transcriptional silencing on sequences located in close proximity (Baur et al. 2001; Karpen and Spradling 1992; Palladino et al. 1993; Schoeftner and Blasco 2009; Wallrath and Elgin 1995). This unique chromatin structure protects the chromosome ends, and loss of this capping function can lead to aneuploidy as chromosome bridges, chromosome fusions, and lagging chromosomes form that will impair mitosis and meiosis (O'Sullivan and Karlseder 2010). In addition, telomeres present a solution to the end-replication problem, which arises because DNA polymerases cannot fully replicate the ends of the lagging strand during DNA replication (de Lange 2009). Ultimately, telomeres perform several functions critical to maintaining genomic integrity.
In order to prevent the loss of sequences at the ends of linear chromosomes with each cell division (end-replication problem), telomeres are composed of repetitive DNA sequences, and several cellular pathways exist to prevent shortening of chromosomes (Jafri et al. 2016). While this function of telomeres is evolutionary conserved, the specific mechanism that maintains telomere length differs from species to species. In most eukaryotes, telomerase, a ribonucleoprotein complex with reverse transcriptase ability, can synthesize new telomeric repeats, thus maintaining telomere length (Greider and Blackburn 1987; Wu et al. 2017a). Saccharomyces cerevisiae also has telomerase activity, but alongside the telomerase, it uses recombination to elongate telomeres (Larrivee and Wellinger 2006; Wellinger and Zakian 2012). In Drosophila melanogaster, telomere length is maintained by two non-LTR retrotransposons, HeT-A and TART, that transpose specifically to chromosome ends (Pardue et al. 2005). Failure of the mechanisms maintaining telomere length leads to shortening telomeres and eventually loss of crucial genetic information located adjacent to the telomeres and cell death (Muraki et al. 2012). Shortening of telomeres is a noteworthy concern in rapidly dividing cells, illustrated by the fact that many cancer cells express high levels of telomerase, which promotes further cell division and growth (Jafri et al. 2016). Thus, telomere maintenance is an essential process to ensure genome stability, and epigenetic mechanisms are required to achieve this goal.
Epigenetic processes can contribute to telomere maintenance by controlling the expression of telomerase in mammals and the expression of the telomeric TEs in insects like Drosophila. In addition, epigenetic modifications, specifically histone modifications, are crucial for controlling telomere length (Counter et al. 1992; Jezek and Green 2019). Changes in telomere chromatin state can alter telomere length and thus lead to genome instability (O'Sullivan and Karlseder 2010). For example, Galán and colleagues found that in yeast, Sus1, a protein involved in histone H2B deubiquitination, is required for the proper regulation of telomere length (Galan et al. 2018). Deletion of Sus1 leads to telomere elongation and an increase in histone H2B lysine 123 (H2BK123) mono-ubiquitination. Thus, the findings by Galán and colleagues suggest that Sus1 negatively regulates telomere length through its impact on histone ubiquitination, linking telomeres, genomic stability, and histone modifications. In addition, several histone methyltransferases (HMTs) have been linked to telomere maintenance as well. Jezek and colleagues found that the Saccharomyces cerevisiae HMTs Set5 and Set1 are involved in telomere maintenance in yeast, and loss of these proteins leads to improper expression of genes adjacent to the telomeres (Jezek et al. 2017). Also, in yeast, Wu and colleagues found that H2BK123 mono-ubiquitination mediated by Rad6-Bre1 positively regulates telomere replication, leading to lengthened telomeres (Wu et al. 2017b). Thus, histone modifications play an important role in maintaining telomere function and genome stability.
As noted previously, telomeres contain a variety of histone modifications (Jezek and Green 2019). These modifications include heterochromatic marks that can exert their influence beyond the telomeric sequences and silence genes nearby through a process known as telomere position effect (TPE) (Robin et al. 2014). TPE is thought to contribute to the genomic instability that arises as telomeres shorten below their critical length (Robin et al. 2014). Recently, it has been suggested that telomeres also impact gene expression over longer distances, a phenomenon named telomere position effects over long distances (TPE-OLD) (Kim and Shay 2018). In TPE-OLD, telomeres cause heterochromatin spreading through chromatin looping, revealed in a 2013 study by Stadler and colleagues examining the regulation of human DUX4, a candidate gene for facioscapulohumeral muscular dystrophy (Stadler et al. 2013). TPE-OLD was confirmed through 3D-FISH (three-dimensional fluorescent in situ hybridization) that showed that chromosomal looping occurred between genes such as ISG15, DSP, and C1S and their respective telomeric ends (Robin et al. 2014). Interestingly, TPE-OLD also seems to affect TERT, the gene encoding telomerase. Kim and colleagues found that telomere length affected expression and nuclear location of the TERT gene (Kim et al. 2016). When telomeres are long, a chromatin loop formed in the hTERT locus, decreasing its expression. As telomeres shortened, for example with age, this loop disengaged and led to increased production of hTERT mRNA. Alongside that disengagement, DNA methylation and histone modifications changed in the hTERT promoter region, suggesting there is a mechanism for fine-tuning telomerase expression based on telomere length in humans (Kim et al. 2016). Thus, TPE-OLD represents another pathway by which telomeres contribute to genome stability utilizing epigenetic mechanisms to impact the expression levels of telomerase and other essential genes.
There are additional chromatin proteins in play that aid in protecting telomere length and function. Shelterin is a complex of chromatin proteins found in eukaryotes that is essential for the chromosome capping function of telomeres, preventing telomeres from fusing with each other (de Lange 2005). It allows telomeres to be distinguished from damaged DNA and specifically binds to chromosome ends, leading to the formation of a unique chromatin structure (de Lange 2005). Eukaryotic shelterin is composed of six core protein subunits: TRF1, TRF2, TIN2, Rap1, TPP1, and POT1 (Xin et al. 2008). Shelterin protects telomeric sites both by binding to the telomeric DNA and by remodeling the chromatin into a unique, tight nucleoprotein structure (Bandaria et al. 2016). TIN2 was the most important component for the shelterin-mediated chromatin compaction of these telomeric sites which prevented the binding of DNA damage response protein, illustrating the importance of chromatin structure at the telomere (Bandaria et al. 2016). Interestingly, the shelterin proteins do not function solely at the telomeres: Loss of TRF2 impairs replication fork progression through pericentromeric heterochromatin and destabilizes heterochromatin across the genome (Mendez-Bermudez et al. 2018), demonstrating that TRF2 has non-telomeric functions as well. Thus, the shelterin protein complex protects telomeres by generating a unique local chromatin structure, illustrating the importance of epigenetic mechanisms for the maintenance of genome stability.
Besides the shelterin proteins, other aspects of chromatin structure also impact the ability of the telomere to carry out its functions. Because the telomeric chromatin is typically considered heterochromatic, Chow and colleagues investigated the effect of increasing the level of the heterochromatin protein HP1α specifically at the telomeres (Chow et al. 2018). To achieve this goal, they fused HP1α with the shelterin protein TRF1, thus altering the telomere chromatin composition. Recruiting TRF1-HP1α fusion protein to the telomere increased heterochromatin formation, altered the 3D structure of the telomere, and prevented access by telomerase, suggesting both positive and negative impacts on telomere function (Chow et al. 2018). This study illustrates the importance of chromatin structure for telomere function and again links an epigenetic system with genome instability.
All in all, the data available today demonstrate that telomeres are unique chromatin structures maintained through epigenetic modifications, which serve to protect the ends of linear chromosomes, thus ensuring genome stability. Loss of telomere function or shortening of telomeres in healthy cells can lead to genome instability, and this loss is regulated by epigenetic pathways including the shelterin chromatin complex (de Lange 2005) and various histone modifications (Jezek and Green 2019). The telomere itself can contribute epigenetically to genome stability—or instability once shortened—through TPE (Robin et al. 2014; Stadler et al. 2013) and the TPE-OLD effect (Kim et al. 2016; Kim and Shay 2018). Interestingly, maintenance of telomere length in cancerous cells through epigenetic alterations leads to telomerase activity that lengthens and maintains telomere length in cancerous cells, aiding in the proliferation of these unstable cells (Jafri et al. 2016; Maciejowski and de Lange 2017). In conclusion, there are various epigenetic factors ensuring functional telomeres that can contribute to genome stability in complex ways.
Epigenetic mechanisms curb TE activity to prevent genome instability
TEs are another common source of genome instability (Klein and O'Neill 2018). TEs are mobile genetic elements that are classified based on the mechanism used for transposition: DNA transposons utilize a DNA intermediate, while RNA transposons or retrotransposons utilize an RNA intermediate (Mc 1950; Padeken et al. 2015). Together, various classes of TEs contribute large amounts of DNA to eukaryotic genomes. For example, TE-derived sequences make up approximately 50% of the human genome (Platt et al. 2018), they contribute approximately 20% to the Drosophila melanogaster genome (McCullers and Steiniger 2017), and in maize, an estimated 85% of the genome sequence is derived from TEs (Jiao et al. 2017; Schnable et al. 2009). While TEs are found throughout the genome, often they are concentrated in specific regions of genomes such as the centromeric and telomeric regions, where they contribute to the function of these domains (see above). Given the large number of TEs in eukaryotes, they are an important facet of these genomes, and epigenetic mechanisms control their activity to ensure genome stability.
Because TEs are able to move, either from location to location or by inserting additional copies of themselves at new locations, they can be a major source of genome instability. With every move, TEs potentially can disrupt genes or gene regulatory pathways. For example, in their review from 2012, Hancks and Kazazian identify 96 retrotransposon insertions that lead to genetic diseases in humans, including cases of hemophilia A and B and cystic fibrosis (Hancks and Kazazian 2012). Given the large number of TEs in many genomes, the mutation rate due to TE transposition can be magnitudes higher than the mutation rate due to mistakes during DNA replication. In addition, TE-encoded endonucleases can cause DNA double-strand breaks without TE insertions, thus potentially causing mutations due to imperfect break repair (Gasior et al. 2006). Finally, TEs contribute to genome instability by facilitating ectopic recombination/unequal crossovers between non-homologous sites due to the presence of identical sequences at various locations in the genome (Cordaux and Batzer 2009). These ectopic recombination events can lead to local deletions or duplications, chromosomal translocations, and inversions. These events have been reported in patients [as in a 2017 case of mesomelia-synostosis syndrome (Kohmoto et al. 2017)] as well as in model organisms. For example, in Drosophila melanogaster, when a DNA transposon-derived reporter gene (P element) is mobilized, local sequence duplications and deletions as well as more complex rearrangements result (Berg et al. 1980; Riddle et al. 2008; Sun et al. 2004). These examples illustrate how TEs contribute to genome instability and why they are targeted for silencing to keep their activity in check.
Given the challenge to genome stability provided by the large number of TEs present in typical eukaryotic genomes, it is not surprising that a variety of mechanisms are in place to curb TE activity (Klein and O'Neill 2018; Molaro and Malik 2016). Several of the TE silencing mechanisms are epigenetic in nature, and both DNA and histone modifications are involved in the transcriptional silencing of TEs, while a number of small RNA pathways are involved in the transcriptional and post-transcriptional silencing of TEs (Dumesic and Madhani 2014). In addition, there are several other pathways, including one linked to p53 (Levine et al. 2016; Tiwari et al. 2018) and one linked to the KAP1 and ZNF transcription factors (Friedli and Trono 2015) that have been implicated in TE silencing. This variety of parallel silencing mechanism hints at the importance of TE silencing for an organism’s survival.
DNA modifications are one level of epigenetic control involved in the control of TE activity (Deniz et al. 2019). DNA methylation in the form of cytosine methylation, in most eukaryotes, is considered a silencing mark, and high levels of cytosine methylation in promoter regions lead to gene silencing and the recruitment of additional silencing chromatin marks in the form of histone modifications (Jones 2012). The majority of TEs show high levels of cytosine methylation and are typically transcriptionally inactive other than in specific tissues where silencing mechanisms are suspended temporarily (Deniz et al. 2019). The importance of DNA methylation for TE silencing and genome stability is illustrated best by what happens if the DNA methylation pathway is impaired. In this case, TE activity, including TE expression levels as well as transpositions, increases. For example, in Arabidopsis thaliana, loss of the SWI2/SNF2 chromatin remodeling factor DDM1 (Jeddeloh et al. 1999), which is required for wildtype levels of cytosine methylation, leads to the reactivation of TEs such as the CAC family (Miura et al. 2001), with increased expression and transposition rate. In maize, with its more than 85% of the genome being derived from TEs, loss of its two DDM1 orthologs is lethal, suggesting that loss of control of a large number of TEs leads to fatal genome instability (Li et al. 2014). In mice with impaired function of the DNA methyltransferase DNMT1, hypomethylation leads to increased somatic transposition of the retroviral-like intracisternal A particle (IAP) (Howard et al. 2008). A 2018 study from Arabidopsis thaliana directly demonstrated that targeted demethylation of the CACTA1 transposon via a CRISPR/dCas9 derived TET1 fusion enzyme increased expression from this TE significantly (Gallego-Bartolome et al. 2018). Together, these studies demonstrate the importance of DNA modifications such as cytosine methylation in ensuring eukaryotic genome stability by silencing TEs.
Along with DNA methylation, histone modifications and chromatin structure, specifically heterochromatin, also contribute to the maintenance of genome stability by silencing TEs (Janssen et al. 2018). Many TEs are found in heterochromatic regions of the genome, typically the centromeres and telomeres, which are characterized by methylation of histone 3 lysine 9 (H3K9me2 and me3) and heterochromatin proteins such as those of the Heterochromatin Protein 1 (HP1) family (Janssen et al. 2018). The chromatin structure in heterochromatin is such that it often suppresses gene expression, leading to the silencing of TEs present in this genomic domain. Biochemical marks of heterochromatin are found also at many TEs within broader euchromatic regions of genomes, which again promote their transcriptional silencing. In Drosophila melanogaster, ~ 30% of euchromatic TEs are associated with silencing marks, and at many of these sites, heterochromatin spreads to neighboring sequences (Lee and Karpen 2017; Riddle et al. 2011). A 2018 screen in human cell lines revealed that transposition of LINE-1 (long interspersed element-1) was controlled in part by the human silencing hub (HUSH) complex, a silencing complex that functions through H3K9 methylation (Liu et al. 2018). The SETDB1 H3K9 methyltransferase was another hit in this screen (Liu et al. 2018), confirming the important role of histone modifications and particularly H3K9 methylation in the silencing of TEs. When He and colleagues screen 41 chromatin modifiers for their impact on TE expression, 29 of the modifiers impacted the expression levels of at least one TE class, and six modifiers, including SETDB1, impacted most TEs (He et al. 2019). Thus, the available data regarding histone modifications and TE activity suggest that repressive histone marks and heterochromatin formation are targeted purposefully to TEs to decrease their expression and ensure genome stability.
In addition, small RNA pathways play an important role in controlling TE activity, as these pathways are involved in targeting specific sequences for DNA modifications and/or heterochromatin formation (Dumesic et al. 2013) (Fig. 2). In plants, the RNA-directed DNA methylation (RdDM) pathway functions via small RNAs to target specific DNA sequences, including TEs, for modification by DNA methyltransferases (Cuerda-Gil and Slotkin 2016; Matzke and Mosher 2014). This pathway relies on a specialized Argonaute protein as well as two plant-specific homologs of RNA pol II (Pol IV and Pol V) to produce small RNAs (siRNAs) complementary to the sequences targeted for DNA methylation (Wendte and Pikaard 2017; Zhou and Law 2015). Defects in this pathway, which has several subtypes, lead to altered chromatin structure and loss of TE silencing (Cuerda-Gil and Slotkin 2016). For example, in Arabidopsis thaliana, HDA6 and PolIV/V cooperatively silence TEs, and the loss of either component leads to TE reactivation, albeit with slightly different effects (Blevins et al. 2014). In animals, piRNAs are the main non-coding RNA type involved in TE control (for a review of their role in mammals, see Ernst et al. (2017); Fig. 2b). The main function of piRNAs is in the germline, where several downstream pathways are important for the TE silencing. piRNAs are derived from long transcripts of so-called piRNA cluster, which can be considered “graveyards” of TEs. The piRNAs then interact with a Piwi-class Argonaute protein, and they are reported to mediate both transcriptional and post-transcriptional silencing of TEs (Czech et al. 2018; Ernst et al. 2017; Hirakata and Siomi 2019; Huang et al. 2017; Ozata et al. 2019; Sentmanat et al. 2013). Failure of the piRNA pathway to silence TEs is illustrated well by the phenomenon of hybrid dysgenesis in Drosophila (Castro and Carareto 2004; Malone et al. 2015). If a strain with the P element (P strain), a DNA transposon which invaded wild Drosophila after the collection of most laboratory strains, is crossed to a laboratory strain lacking P elements (M strain), the P elements are activated in the F1 germline, and the animals exhibit various defects including sterility (Kidwell et al. 1977). This sterility can be prevented by the presence of piRNAs to the TE in the F1 germline, which occurs naturally in the reciprocal cross, which does not lead to dysgenesis (Brennecke et al. 2008). Mutants in the piRNA pathways generally lead to sterility and problems in the germline, and in many aspects, these animals resemble dysgenic animals (Kelleher et al. 2012). Together, the experiments investigating small RNA pathways in animals and plants illustrate the importance of these pathways for the formation of silent chromatin, the control of TEs, and genome stability.

Examples of small RNA-mediated TE silencing. a RdDM pathway in Arabidopsis thaliana. RNA Polymerase IV (Pol IV) produces a single-stranded transcript from a transposon (i). RNA-dependent RNA Polymerase 2 (RDR2) uses this single-stranded RNA as a template to produce a double-stranded RNA (ii). This double-stranded RNA is diced into 24-nt small interfering RNAs (siRNAs) by Dicer-like 3 (DCL3) (iii). AGO4 binds the 24-nt siRNA. Next, this complex binds to matching Pol V-generated transcripts, still associated with their transcription unit (iv). This binding recruits the domains of rearranged methyltransferase 2 (DRM2), which produces cytosine methylation and silences the targeted transposon. Pol IV continues to produce transposon transcripts at a low level, feeding back into the siRNA producing pathway (v). b The piRNA pathway in Drosophila melanogaster. A piRNA precursor is transcribed from a piRNA cluster containing TE sequences and exported from the nucleus to the cytoplasm (i). The piRNA precursor then is processed into small, ~ 24 bp-length piRNAs by the Zucchini protein (Zuc, red) at the mitochondria (ii), which can be bound to PIWI and translocated into the nucleus to transcriptionally silence homologous TEs (iii). Alternatively, piRNA precursor transcripts also can be processed by the ping-pong cycle (v). In the ping-pong amplification cycle, a mature piRNA bound to the argonaute protein Aubergine (AUB) (iv) directs slicing of a matching transposon transcript bound to AGO3, which will be processed into a mature piRNA. The newly formed AGO3-piRNA complex can then slice additional matching sequences, either precursor piRNAs or transposon transcripts, which will bind to AUB (v). Continuation of this cycle leads to amplification of piRNAs, which can either be utilized in the ping-pong cycle to post-transcriptionally degrade transposon transcripts or bound to PIWI and translocated into the nucleus to transcriptionally silence homologous TEs (III)
In summary, while having some beneficial functions at the centromere, telomere, and in generating evolutionary novelty, TEs are a major source of genome instability due to their ability to jump from locus to locus and due to the consequences of the presence of numerous identical sequences at non-homologous locations for other nuclear processes (DNA repair, replication, etc.). Thus, epigenetic processes working through DNA modifications, histone modifications, chromatin structure, and small RNAs are essential to promoting genome stability by ensuring the suppression of TE expression and transposition. Failure of these pathways to suppress TEs has severe consequences, including sterility and even death in species with high levels of TEs, highlighting the importance of the epigenetic processes involved for genome stability.
Epigenetic modifications in DNA repair pathways impact genome stability
As noted in the section on TEs, DNA repair is an important aspect of genome stability (Lombard et al. 2005). The genome is experiencing constant assault from both internal sources such as TEs and from external factors such as chemical mutagens, UV radiation, and more (Chatterjee and Walker 2017). Different DNA damage repair pathways exist to deal with different types of damage. One of the most common forms of DNA damage are DNA double-stranded breaks (DSB), such as the breaks introduced by the transposition of TEs (for example, see Liu and Wessler 2017). DSBs can be repaired through mechanisms such as non-homologous end joining (NHEJ) and homologous recombination (HR), which uses homologous sequences as a repair template (Brandsma and van Gent 2012). Abnormal bases or pyrimidine dimers created for example by UV radiation are corrected through the nucleotide excision repair (NER) or base excision repair (BER) pathways (Melis et al. 2013). The DNA mismatch repair pathway typically corrects mistakes introduced during DNA replication, and direct reversal repair pathways are available for some altered bases (Chatterjee and Walker 2017). In many cancers, increased genome instability is seen, which has been traced back to the inactivation of genes involved in various DNA repair pathways (Lahtz and Pfeifer 2011). This finding illustrates that DNA repair pathways are crucial for maintaining genomic stability by preventing DNA damage from accumulating and negatively impacting cell functions.
Of the four core histones, H2A has the largest number of variants studied to date, and one of these variants, H2AX is linked to DNA repair and genome stability (Georgoulis et al. 2017). In mammalian cells, H2AX is a histone variant of H2A that is rapidly phosphorylated with high specificity in response to DSBs, yielding γ-H2AX (Firsanov et al. 2011; Kinner et al. 2008; Rogakou et al. 1998). Phosphorylation is removed from H2AX after the DSB has been repaired and chromatin integrity has been restored. Celeste and colleagues investigated cells with reduced H2AX levels and found that H2AX is not crucial for initial recognition of the DSBs; however, cells without H2AX were less effective at recruiting DNA damage response proteins later in the repair process (Celeste et al. 2003). Ultimately, γ-H2AX’s function appears to be to modify the chromatin structure, to increase DNA accessibility, and to recruit DNA damage response proteins to the appropriate regions in the genome (Celeste et al. 2002). Consequently, γ-H2AX has become a noteworthy molecular marker for DNA damage and is a hallmark of genomic instability. Thus, γ-H2AX might serve as a prognostic biomarker for various cancers, such as breast (Nagelkerke et al. 2011; Varvara et al. 2019; Wang et al. 2019), lung (Chatzimichail et al. 2014; Matthaios et al. 2012; Ochola et al. 2019), and bladder cancers (Fernández et al. 2013). Together, the available data demonstrate that γ-H2AX is critical for genome integrity due to its role in the DNA DSB repair pathway, highlighting another route by which epigenetics contributes to genome stability.
An additional group of epigenetic regulators of DNA repair pathways is represented by the sirtuin protein family. Sirtuins are highly conserved proteins with histone deacetylase functions, occurring in species from bacteria to yeast to humans (Greiss and Gartner 2009; Imai et al. 2000). Sir2 from budding yeast was the first sirtuin to be described, and in this species, it is essential for gene silencing in heterochromatin regions including the telomeres (Aparicio et al. 1991; Rine and Herskowitz 1987; Wierman and Smith 2014). Sirtuins have been linked to a variety of biological processes, including aging, transcription, and stress response, and it is often their role in DNA repair mechanisms that influences these processes (Choi and Mostoslavsky 2014). Among the mammalian sirtuins, SIRT1 is the most well studied, and SIRT1 has been linked to DNA repair pathways including the HR DNA repair pathway, the NHEJ DNA repair pathway, and the NER pathway (Fan and Luo 2010; Li et al. 2008; Yamamori et al. 2010). Lin and colleagues recently found that SIRT1 is involved in the choice between the HR and NHEJ repair pathways for DNA DSBs (Lin et al. 2015). They demonstrated that KAP1, a transcriptional corepressor, inhibits HR- and promotes NHEJ-mediated repair of DSBs, but that this process depends on the KAP1 acetylation state. Deacetylation of KAP1 is regulated by SIRT1, and it is this deacetylation that promotes NHEJ-mediated repair (Lin et al. 2015). In addition to KAP1, SIRT1 also deacetylates KU70, a DNA damage repair protein that is essential to the NHEJ pathway (Fell and Schild-Poulter 2012). Zhang et al. found that SIRT1 enhances NHEJ activity in chronic myeloid leukemia (CML) cells, likely through its role in deacetylating KU70 (Zhang et al. 2016). In addition, Roth and colleagues found that SIRT1 and the LSD1 lysine demethylase competitively bind to KU70 in cancer cells responding to stress, having different effects on NHEJ repair efficiency (Roth et al. 2016). Thus, SIRT1 is an example of an epigenetic modifier which impacts the NHEJ repair pathway, thus contributing to the control of genome stability.
The roles of histone variants like H2AX and epigenetic modifiers like SIRT1 and LSD1 in DNA repair pathways illustrate another important link between epigenetics and genome stability. These examples show that DNA repair pathways utilize a variety of epigenetic mechanisms combating DNA damage to ensure genome stability, preventing aging and cancer. All in all, these are just a few of the epigenetic mechanisms of note that impact DNA repair pathways and genomic stability.
Conclusions and outlook
As illustrated in this review, various epigenetic mechanisms are at work in maintaining genome stability. Epigenetic systems regulate centromere organization to ensure proper chromosome segregation during cell division, and their loss can upset genomic integrity and lead to chromosomal abnormalities and cell death (Barra and Fachinetti 2018). Without the correct epigenetic mediators, telomeres can shorten, or lose their ability to protect chromosome ends (Booth and Brunet 2016; Maciejowski and de Lange 2017; O'Sullivan and Karlseder 2010). Without the epigenetic mechanisms controlling their activity, TEs can cause genomic instability by disrupting essential genes, introducing DNA DSBs, and promoting errors during DNA replication, DNA repair, and homologous recombination (Klein and O'Neill 2018). Epigenetic mechanisms also are essential regulators of the DNA repair pathways that are core to maintaining genome stability in the face of DNA damage, and likely contribute to other pathways contributing to genome stability as well (Dabin et al. 2016). Thus, epigenetic mechanisms are an integral part of the complex web of pathways used to ensure genome stability.
While the basic fact of the involvement of epigenetic systems in the regulation of genome stability is well-established, details for many pathways are still lacking and are areas of active research. Epigenetic mechanisms likely will play many additional roles in the gene regulatory pathways that control the expression of the large variety of proteins and transcripts that contribute to genome stability. In addition, there is an interesting but currently limited body of research looking into the question of how the epigenome, in particular, chromatin structure influences the mutational landscape. These studies indicate that mutation rates differ between regions of the genome with well-positioned nucleosomes and regions that are nucleosome-free, with nucleosomal DNA surprisingly showing higher levels of substitutions and regions of DNA typically found in the linker between nucleosomes (Warnecke et al. 2008; Washietl et al. 2008). Additionally, there is some data suggesting that not all nucleosomes impact substitution rates equally and that the histone modifications present on a particular nucleosome might affect mutation rates (Prendergast and Semple 2011; Tolstorukov et al. 2011). The question of how chromatin structure impacts the mutations that occur and the efficiency of the DNA repair pathways is highly relevant and ripe for further exploration.
In addition, studies using a comparative approach are likely to shed more light onto the connections between epigenetic mechanisms and genome stability. The progress in sequencing technologies and the drop in cost have made genomics and epigenomics studies possible in any species of interest. The resulting increase in datasets from a large variety of species has led, for example, to the realization that not all species use CenH3 to determine centromere identity (Akiyoshi and Gull 2014; Berriman et al. 2005; Drinnenberg et al. 2014) and raises the question of how the centromere works in these species. Experiences like this one suggest that there are many more novel links between epigenetics and genome stability awaiting their discovery. Especially histone variants, telomere structure, and centromeres are promising areas of further research, because the variation present among well-characterized species suggests that more is likely to be found once a wider range of species is considered.
References
Aguilera A, Garcia-Muse T (2013) Causes of genome instability. Annu Rev Genet 47:1–32. https://doi.org/10.1146/annurev-genet-111212-133232
Akiyoshi B, Gull K (2014) Discovery of unconventional kinetochores in kinetoplastids. Cell 156:1247–1258. https://doi.org/10.1016/j.cell.2014.01.049
Aldrup-MacDonald ME, Kuo ME, Sullivan LL, Chew K, Sullivan BA (2016) Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res 26:1301–1311. https://doi.org/10.1101/gr.206706.116
Amor DJ, Bentley K, Ryan J, Perry J, Wong L, Slater H, Choo KHA (2004) Human centromere repositioning "in progress". Proc Natl Acad Sci USA 101:6542–6547. https://doi.org/10.1073/pnas.0308637101
Aparicio OM, Billington BL, Gottschling DE (1991) Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell 66:1279–1287. https://doi.org/10.1016/0092-8674(91)90049-5
Bandaria JN, Qin P, Berk V, Chu S, Yildiz A (2016) Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 164:735–746. https://doi.org/10.1016/j.cell.2016.01.036
Barra V, Fachinetti D (2018) The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat Commun 9:4340. https://doi.org/10.1038/s41467-018-06545-y
Barra V et al (2019) Phosphorylation of CENP-A on serine 7 does not control centromere function. Nature Commun 10:175. https://doi.org/10.1038/s41467-018-08073-1
Baur JA, Zou Y, Shay JW, Wright WE (2001) Telomere position effect in human cells. Science 292:2075–2077. https://doi.org/10.1126/science.1062329
Berg R, Engels WR, Kreber RA (1980) Site-specific X-chromosome rearrangements from hybrid dysgenesis in Drosophila melanogaster. Science 210:427–429. https://doi.org/10.1126/science.6776625
Bergmann JH et al (2012) Epigenetic engineering: histone H3K9 acetylation is compatible with kinetochore structure and function. J Cell Sci 125:411–421. https://doi.org/10.1242/jcs.090639
Berriman M et al (2005) The genome of the African trypanosome Trypanosoma brucei. Science 309:416–422. https://doi.org/10.1126/science.1112642
Blevins T et al (2014) A two-step process for epigenetic inheritance in Arabidopsis. Mol Cell 54:30–42. https://doi.org/10.1016/j.molcel.2014.02.019
Blower MD, Sullivan BA, Karpen GH (2002) Conserved organization of centromeric chromatin in flies and humans. Dev Cell 2:319–330. https://doi.org/10.1016/s1534-5807(02)00135-1
Booth LN, Brunet A (2016) The aging epigenome. Mol Cell 62:728–744. https://doi.org/10.1016/j.molcel.2016.05.013
Bowman GD, Poirier MG (2015) Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev 115:2274–2295. https://doi.org/10.1021/cr500350x
Brandsma I, van Gent DC (2012) Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integrity 3:9. https://doi.org/10.1186/2041-9414-3-9
Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ (2008) An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 322:1387–1392. https://doi.org/10.1126/science.1165171
Brenner M, Hearing VJ (2008) The protective role of melanin against UV damage in human skin. Photochem Photobiol 84:539–549. https://doi.org/10.1111/j.1751-1097.2007.00226.x
Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O'Geen H, Cotterman RF, Knoepfler PS (2013) Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 6:7. https://doi.org/10.1186/1756-8935-6-7
Caneus J, Granic A, Rademakers R, Dickson DW, Coughlan CM, Chial HJ, Potter H (2018) Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol Biol Cell 29:575–586. https://doi.org/10.1091/mbc.E17-01-0031
Castro JP, Carareto CM (2004) Drosophila melanogaster P transposable elements: mechanisms of transposition and regulation. Genetica 121:107–118. https://doi.org/10.1023/b:gene.0000040382.48039.a2
Celeste A et al (2002) Genomic instability in mice lacking histone H2AX. Science 296:922–927. https://doi.org/10.1126/science.1069398
Celeste A et al (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5:675–679. https://doi.org/10.1038/ncb1004
Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506. https://doi.org/10.1038/nrm.2017.48
Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58:235–263. https://doi.org/10.1002/em.22087
Chatzimichail E et al (2014) gamma -H2AX: A novel prognostic marker in a prognosis prediction model of patients with early operable non-small cell lung cancer. Int J Genomics 2014:160236. https://doi.org/10.1155/2014/160236
Choi J-E, Mostoslavsky R (2014) Sirtuins, metabolism, and DNA repair. Curr Opin Genet Dev 26:24–32. https://doi.org/10.1016/j.gde.2014.05.005
Chow TT, Shi X, Wei JH, Guan J, Stadler G, Huang B, Blackburn EH (2018) Local enrichment of HP1alpha at telomeres alters their structure and regulation of telomere protection. Nat Commun 9:3583. https://doi.org/10.1038/s41467-018-05840-y
Choy JS, Mishra PK, Au W-C, Basrai MA (2012) Insights into assembly and regulation of centromeric chromatin in Saccharomyces cerevisiae. Biochim Biophys Acta 1819:776–783. https://doi.org/10.1016/j.bbagrm.2012.02.008
Chunduri NK, Storchova Z (2019) The diverse consequences of aneuploidy. Nat Cell Biol 21:54–62. https://doi.org/10.1038/s41556-018-0243-8
Cohen AK, Huh TY, Helleiner CW (1973) Transcription of satellite DNA in mouse L-cells. Can J Biochem 51:529–532. https://doi.org/10.1139/o73-065
Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10:691–703. https://doi.org/10.1038/nrg2640
Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11:1921–1929
Cuacos M, Franklin FC H, Heckmann S (2015) Atypical centromeres in plants-what they can tell us. Front Plant Sci 6:913–913. https://doi.org/10.3389/fpls.2015.00913
Cuerda-Gil D, Slotkin RK (2016) Non-canonical RNA-directed DNA methylation. Nat Plants 2:16163. https://doi.org/10.1038/nplants.2016.163
Czech B, Munafo M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, Hannon GJ (2018) piRNA-guided genome defense: from biogenesis to silencing. Annu Rev Genet 52:131–157. https://doi.org/10.1146/annurev-genet-120417-031441
Dabin J, Fortuny A, Polo SE (2016) Epigenome maintenance in response to DNA damage. Mol Cell 62:712–727. https://doi.org/10.1016/j.molcel.2016.04.006
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110. https://doi.org/10.1101/gad.1346005
de Lange T (2009) How telomeres solve the end-protection problem. Science (New York, NY) 326:948–952. https://doi.org/10.1126/science.1170633
Deniz O, Frost JM, Branco MR (2019) Regulation of transposable elements by DNA modifications. Nat Rev Genet 20:417–431. https://doi.org/10.1038/s41576-019-0106-6
Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ (2007) Dynamics of replication-independent histone turnover in budding yeast. Science 315:1405–1408. https://doi.org/10.1126/science.1134053
Drinnenberg IA, deYoung D, Henikoff S, Malik HS (2014) Recurrent loss of CenH3 is associated with independent transitions to holocentricity in insects. eLife 3:e03676. https://doi.org/10.7554/eLife.03676
Drinnenberg IA, Henikoff S, Malik HS (2016) Evolutionary turnover of kinetochore proteins: a ship of theseus? Trends Cell Biol 26:498–510. https://doi.org/10.1016/j.tcb.2016.01.005
Dumesic PA, Madhani HD (2014) Recognizing the enemy within: licensing RNA-guided genome defense. Trends Biochem Sci 39:25–34. https://doi.org/10.1016/j.tibs.2013.10.003
Dumesic PA et al (2013) Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152:957–968. https://doi.org/10.1016/j.cell.2013.01.046
Ernst C, Odom DT, Kutter C (2017) The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun 8:1411. https://doi.org/10.1038/s41467-017-01049-7
Fan W, Luo J (2010) SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell 39:247–258. https://doi.org/10.1016/j.molcel.2010.07.006
Fell VL, Schild-Poulter C (2012) Ku regulates signaling to DNA damage response pathways through the Ku70 von Willebrand A domain. Mol Cell Biol 32:76–87. https://doi.org/10.1128/MCB.05661-11
Felsenfeld G (2014) A brief history of epigenetics. Cold Spring Harb Perspect Biol 6:a018200. https://doi.org/10.1101/cshperspect.a018200
Fernández MI, Gong Y, Ye Y, Lin J, Chang DW, Kamat AM, Wu X (2013) γ-H2AX level in peripheral blood lymphocytes as a risk predictor for bladder cancer. Carcinogenesis 34:2543–2547. https://doi.org/10.1093/carcin/bgt270
Firsanov DV, Solovjeva LV, Svetlova MP (2011) H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin Epigenetics 2:283–297. https://doi.org/10.1007/s13148-011-0044-4
Fischer KE, Riddle NC (2018) Sex differences in aging: genomic instability. J Gerontol A 73:166–174. https://doi.org/10.1093/gerona/glx105
Franklin SG, Zweidler A (1977) Non-allelic variants of histones 2a, 2b and 3 in mammals. Nature 266:273–275. https://doi.org/10.1038/266273a0
Friedli M, Trono D (2015) The developmental control of transposable elements and the evolution of higher species. Annu Rev Cell Dev Biol 31:429–451. https://doi.org/10.1146/annurev-cellbio-100814-125514
Fukagawa T, Earnshaw WC (2014a) The centromere: chromatin foundation for the kinetochore machinery. Dev Cell 30:496–508. https://doi.org/10.1016/j.devcel.2014.08.016
Fukagawa T, Earnshaw WC (2014b) Neocentromeres. Curr Biol 24:R946–947. https://doi.org/10.1016/j.cub.2014.08.032
Furuyama S, Biggins S (2007) Centromere identity is specified by a single centromeric nucleosome in budding yeast. Proc Natl Acad Sci USA 104:14706–14711. https://doi.org/10.1073/pnas.0706985104
Galan A, Garcia-Oliver E, Nuno-Cabanes C, Rubinstein L, Kupiec M, Rodriguez-Navarro S (2018) The evolutionarily conserved factor Sus1/ENY2 plays a role in telomere length maintenance. Curr Genet 64:635–644. https://doi.org/10.1007/s00294-017-0778-4
Gallego-Bartolome J et al (2018) Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc Natl Acad Sci USA 115:E2125–E2134. https://doi.org/10.1073/pnas.1716945115
Gambogi Craig W, Black Ben E (2019) The nucleosomes that mark centromere location on chromosomes old and new. Essays Biochem 63:15–27. https://doi.org/10.1042/EBC20180060
Gasior SL, Wakeman TP, Xu B, Deininger PL (2006) The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357:1383–1393. https://doi.org/10.1016/j.jmb.2006.01.089
Gent JI, Wang N, Dawe RK (2017) Stable centromere positioning in diverse sequence contexts of complex and satellite centromeres of maize and wild relatives. Genome Biol 18:121. https://doi.org/10.1186/s13059-017-1249-4
Georgoulis A, Vorgias CE, Chrousos GP, Rogakou EP (2017) Genome Instability and gammaH2AX. Int J Mol Sci. https://doi.org/10.3390/ijms18091979
Greider CW, Blackburn EH (1987) The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51:887–898. https://doi.org/10.1016/0092-8674(87)90576-9
Greiss S, Gartner A (2009) Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol Cells 28:407–415. https://doi.org/10.1007/s10059-009-0169-x
Hall IM, Shankaranarayana GD, Noma K, Ayoub N, Cohen A, Grewal SI (2002) Establishment and maintenance of a heterochromatin domain. Science 297:2232–2237. https://doi.org/10.1126/science.1076466
Hancks DC, Kazazian HH Jr (2012) Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22:191–203. https://doi.org/10.1016/j.gde.2012.02.006
He J et al (2019) Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat Commun 10:34. https://doi.org/10.1038/s41467-018-08006-y
Henikoff S, Furuyama T (2012) The unconventional structure of centromeric nucleosomes. Chromosoma 121:341–352. https://doi.org/10.1007/s00412-012-0372-y
Henikoff S, Smith MM (2015) Histone variants and epigenetics. Cold Spring Harb Perspect Biol 7:a019364–a019364. https://doi.org/10.1101/cshperspect.a019364
Hirakata S, Siomi MC (2019) Assembly and function of gonad-specific non-membranous organelles in Drosophila piRNA biogenesis. Noncoding RNA. https://doi.org/10.3390/ncrna5040052
Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A (2008) Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 27:404–408. https://doi.org/10.1038/sj.onc.1210631
Huang X, Fejes Toth K, Aravin AA (2017) piRNA biogenesis in Drosophila melanogaster. Trends Genet 33:882–894. https://doi.org/10.1016/j.tig.2017.09.002
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800. https://doi.org/10.1038/35001622
Jafri MA, Ansari SA, Alqahtani MH, Shay JW (2016) Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med 8:69. https://doi.org/10.1186/s13073-016-0324-x
Jang CW, Shibata Y, Starmer J, Yee D, Magnuson T (2015) Histone H3.3 maintains genome integrity during mammalian development. Genes Dev 29:1377–1392. https://doi.org/10.1101/gad.264150.115
Janssen A, Colmenares SU, Karpen GH (2018) Heterochromatin: guardian of the genome. Annu Rev Cell Dev Biol 34:265–288. https://doi.org/10.1146/annurev-cellbio-100617-062653
Jeddeloh JA, Stokes TL, Richards EJ (1999) Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet 22:94–97. https://doi.org/10.1038/8803
Jezek M, Green EM (2019) Histone modifications and the maintenance of telomere integrity. Cells 8:199. https://doi.org/10.3390/cells8020199
Jezek M et al (2017) The histone methyltransferases Set5 and Set1 have overlapping functions in gene silencing and telomere maintenance. Epigenetics 12:93–104. https://doi.org/10.1080/15592294.2016.1265712
Jiao Y et al (2017) Improved maize reference genome with single-molecule technologies. Nature 546:524–527. https://doi.org/10.1038/nature22971
Johnson L, Mollah S, Garcia BA, Muratore TL, Shabanowitz J, Hunt DF, Jacobsen SE (2004) Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res 32:6511–6518. https://doi.org/10.1093/nar/gkh992
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492. https://doi.org/10.1038/nrg3230
Karpen GH, Spradling AC (1992) Analysis of subtelomeric heterochromatin in the Drosophila minichromosome Dp1187 by single P element insertional mutagenesis. Genetics 132:737–753
Kelleher ES, Edelman NB, Barbash DA (2012) Drosophila interspecific hybrids phenocopy piRNA-pathway mutants. PLoS Biol 10:e1001428. https://doi.org/10.1371/journal.pbio.1001428
Kidwell MG, Kidwell JF, Sved JA (1977) Hybrid dysgenesis in DROSOPHILA MELANOGASTER: a syndrome of aberrant traits including mutation, sterility and male recombination. Genetics 86:813–833
Kim W, Shay JW (2018) Long-range telomere regulation of gene expression: Telomere looping and telomere position effect over long distances (TPE-OLD). Differentiation 99:1–9. https://doi.org/10.1016/j.diff.2017.11.005
Kim W et al (2016) Regulation of the human telomerase gene TERT by telomere position effect-over long distances (TPE-OLD): implications for aging and cancer. PLoS Biol 14:e2000016. https://doi.org/10.1371/journal.pbio.2000016
Kinner A, Wu W, Staudt C, Iliakis G (2008) Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 36:5678–5694. https://doi.org/10.1093/nar/gkn550
Klein SJ, O'Neill RJ (2018) Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Res 26:5–23. https://doi.org/10.1007/s10577-017-9569-5
Kohmoto T et al (2017) A 590 kb deletion caused by non-allelic homologous recombination between two LINE-1 elements in a patient with mesomelia-synostosis syndrome. Am J Med Genet A 173:1082–1086. https://doi.org/10.1002/ajmg.a.38122
Lahtz C, Pfeifer GP (2011) Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol 3:51–58. https://doi.org/10.1093/jmcb/mjq053
Larrivee M, Wellinger RJ (2006) Telomerase- and capping-independent yeast survivors with alternate telomere states. Nat Cell Biol 8:741–747. https://doi.org/10.1038/ncb1429
Lee YCG, Karpen GH (2017) Pervasive epigenetic effects of Drosophila euchromatic transposable elements impact their evolution. Elife. https://doi.org/10.7554/eLife.25762
Lens SMA, Medema RH (2019) Cytokinesis defects and cancer. Nat Rev Cancer 19:32–45. https://doi.org/10.1038/s41568-018-0084-6
Levine AJ, Ting DT, Greenbaum BD (2016) P53 and the defenses against genome instability caused by transposons and repetitive elements. BioEssays 38:508–513. https://doi.org/10.1002/bies.201600031
Li K et al (2008) Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem 283:7590–7598. https://doi.org/10.1074/jbc.M709707200
Li W, Prazak L, Chatterjee N, Grüninger S, Krug L, Theodorou D, Dubnau J (2013) Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16:529–531. https://doi.org/10.1038/nn.3368
Li Q et al (2014) Genetic perturbation of the maize methylome. Plant Cell 26:4602–4616. https://doi.org/10.1105/tpc.114.133140
Lin YH, Yuan J, Pei H, Liu T, Ann DK, Lou Z (2015) KAP1 Deacetylation by SIRT1 promotes non-homologous end-joining repair. PLoS ONE 10:e0123935. https://doi.org/10.1371/journal.pone.0123935
Ling YH, Yuen KWY (2019) Point centromere activity requires an optimal level of centromeric noncoding RNA. Proc Natl Acad Sci USA 116:6270–6279. https://doi.org/10.1073/pnas.1821384116
Liu K, Wessler SR (2017) Transposition of Mutator-like transposable elements (MULEs) resembles hAT and Transib elements and V(D)J recombination. Nucleic Acids Res 45:6644–6655. https://doi.org/10.1093/nar/gkx357
Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J (2018) Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553:228–232. https://doi.org/10.1038/nature25179
Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) DNA repair, genome stability, and aging. Cell 120:497–512. https://doi.org/10.1016/j.cell.2005.01.028
Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G (2006) PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell 24:309–316. https://doi.org/10.1016/j.molcel.2006.08.019
Maciejowski J, de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18:175–186. https://doi.org/10.1038/nrm.2016.171
Maggert KA, Karpen GH (2001) The activation of a neocentromere in Drosophila requires proximity to an endogenous centromere. Genetics 158:1615–1628
Maison C et al (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30:329–334. https://doi.org/10.1038/ng843
Malik HS, Henikoff S (2003) Phylogenomics of the nucleosome. Nat Struct Biol 10:882–891. https://doi.org/10.1038/nsb996
Malone CD, Lehmann R, Teixeira FK (2015) The cellular basis of hybrid dysgenesis and Stellate regulation in Drosophila. Curr Opin Genet Dev 34:88–94. https://doi.org/10.1016/j.gde.2015.09.003
Maloney KA, Sullivan LL, Matheny JE, Strome ED, Merrett SL, Ferris A, Sullivan BA (2012) Functional epialleles at an endogenous human centromere. Proc Natl Acad Sci USA 109:13704–13709. https://doi.org/10.1073/pnas.1203126109
Matthaios D et al (2012) gamma-H2AX expression detected by immunohistochemistry correlates with prognosis in early operable non-small cell lung cancer. Onco Targets Ther 5:309–314. https://doi.org/10.2147/OTT.S36995
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15:394–408. https://doi.org/10.1038/nrg3683
Mc CB (1950) The origin and behavior of mutable loci in maize. Proc Natl Acad Sci USA 36:344–355. https://doi.org/10.1073/pnas.36.6.344
McCullers TJ, Steiniger M (2017) Transposable elements in Drosophila. Mob Genet Elements 7:1–18. https://doi.org/10.1080/2159256X.2017.1318201
McKinley KL, Cheeseman IM (2016) The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17:16–29. https://doi.org/10.1038/nrm.2015.5
McKittrick E, Gafken PR, Ahmad K, Henikoff S (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci USA 101:1525–1530. https://doi.org/10.1073/pnas.0308092100
Mehta A, Haber JE (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 6:a016428–a016428. https://doi.org/10.1101/cshperspect.a016428
Melis JPM, van Steeg H, Luijten M (2013) Oxidative DNA damage and nucleotide excision repair. Antioxid Redox Signal 18:2409–2419. https://doi.org/10.1089/ars.2012.5036
Mendez-Bermudez A et al (2018) Genome-wide control of heterochromatin replication by the telomere capping protein TRF2. Mol Cell 70(449–461):e445. https://doi.org/10.1016/j.molcel.2018.03.036
Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T (2001) Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411:212–214. https://doi.org/10.1038/35075612
Molaro A, Malik HS (2016) Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr Opin Genet Dev 37:51–58. https://doi.org/10.1016/j.gde.2015.12.001
Molina O et al (2016) Epigenetic engineering reveals a balance between histone modifications and transcription in kinetochore maintenance. Nat Commun 7:13334–13334. https://doi.org/10.1038/ncomms13334
Muraki K, Nyhan K, Han L, Murnane JP (2012) Mechanisms of telomere loss and their consequences for chromosome instability. Front Oncol 2:135–135. https://doi.org/10.3389/fonc.2012.00135
Nagelkerke A et al (2011) Constitutive expression of γ-H2AX has prognostic relevance in triple negative breast cancer. Radiother Oncol 101:39–45. https://doi.org/10.1016/j.radonc.2011.07.009
Nergadze SG et al (2018) Birth, evolution, and transmission of satellite-free mammalian centromeric domains. Genome Res 28:789–799. https://doi.org/10.1101/gr.231159.117
Ochola DO et al (2019) Persistence of gamma-H2AX foci in bronchial cells correlates with susceptibility to radiation associated lung cancer in mice. Radiat Res 191:67–75. https://doi.org/10.1667/RR14979.1
Ohzeki J-I et al (2016) KAT7/HBO1/MYST2 Regulates CENP-A chromatin assembly by antagonizing Suv39h1-mediated centromere inactivation. Dev Cell 37:413–427. https://doi.org/10.1016/j.devcel.2016.05.006
O'Sullivan RJ, Karlseder J (2010) Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11:171–181. https://doi.org/10.1038/nrm2848
Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD (2019) PIWI-interacting RNAs: small RNAs with big functions. Nat Rev Genet 20:89–108. https://doi.org/10.1038/s41576-018-0073-3
Padeken J, Zeller P, Gasser SM (2015) Repeat DNA in genome organization and stability. Curr Opin Genet Dev 31:12–19. https://doi.org/10.1016/j.gde.2015.03.009
Pal S, Tyler JK (2016) Epigenetics and aging. Sci Adv 2:e1600584. https://doi.org/10.1126/sciadv.1600584
Palladino F, Laroche T, Gilson E, Axelrod A, Pillus L, Gasser SM (1993) SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell 75:543–555. https://doi.org/10.1016/0092-8674(93)90388-7
Palmer DK, O'Day K, Trong HL, Charbonneau H, Margolis RL (1991) Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc Natl Acad Sci USA 88:3734–3738. https://doi.org/10.1073/pnas.88.9.3734
Pardue ML, Rashkova S, Casacuberta E, DeBaryshe PG, George JA, Traverse KL (2005) Two retrotransposons maintain telomeres in Drosophila. Chromosome Res 13:443–453. https://doi.org/10.1007/s10577-005-0993-6
Perea-Resa C, Blower MD (2018) Centromere biology: transcription goes on stage. Mol Cell Biol 38:e00263-e00218. https://doi.org/10.1128/MCB.00263-18
Platt RN 2nd, Vandewege MW, Ray DA (2018) Mammalian transposable elements and their impacts on genome evolution. Chromosome Res 26:25–43. https://doi.org/10.1007/s10577-017-9570-z
Plohl M, Meštrović N, Mravinac B (2014) Centromere identity from the DNA point of view. Chromosoma 123:313–325. https://doi.org/10.1007/s00412-014-0462-0
Prendergast JGD, Semple CAM (2011) Widespread signatures of recent selection linked to nucleosome positioning in the human lineage. Genome Res 21:1777–1787. https://doi.org/10.1101/gr.122275.111
Riddle NC, Leung W, Haynes KA, Granok H, Wuller J, Elgin SC (2008) An investigation of heterochromatin domains on the fourth chromosome of Drosophila melanogaster. Genetics 178:1177–1191. https://doi.org/10.1534/genetics.107.081828
Riddle NC et al (2011) Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21:147–163. https://doi.org/10.1101/gr.110098.110
Rine J, Herskowitz I (1987) Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 116:9–22
Robin JD et al (2014) Telomere position effect: regulation of gene expression with progressive telomere shortening over long distances. Genes Dev 28:2464–2476. https://doi.org/10.1101/gad.251041.114
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. https://doi.org/10.1074/jbc.273.10.5858
Rošić S, Köhler F, Erhardt S (2014) Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J Cell Biol 207:335–349. https://doi.org/10.1083/jcb.201404097
Rosin LF, Mellone BG (2017) Centromeres drive a hard bargain. Trends Genet 33:101–117. https://doi.org/10.1016/j.tig.2016.12.001
Roth M, Wang Z, Chen WY (2016) SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 7:50195–50214. https://doi.org/10.18632/oncotarget.10328
Sansregret L, Vanhaesebroeck B, Swanton C (2018) Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol 15:139–150. https://doi.org/10.1038/nrclinonc.2017.198
Schnable PS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115. https://doi.org/10.1126/science.1178534
Schneider KL, Xie Z, Wolfgruber TK, Presting GG (2016) Inbreeding drives maize centromere evolution. Proc Natl Acad Sci USA 113:E987–E996. https://doi.org/10.1073/pnas.1522008113
Schoeftner S, Blasco MA (2009) A 'higher order' of telomere regulation: telomere heterochromatin and telomeric RNAs. EMBO J 28:2323–2336. https://doi.org/10.1038/emboj.2009.197
Schwartz BE, Ahmad K (2005) Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev 19:804–814. https://doi.org/10.1101/gad.1259805
Scott KC, Sullivan BA (2014) Neocentromeres: a place for everything and everything in its place. Trends Genet 30:66–74. https://doi.org/10.1016/j.tig.2013.11.003
Scully R, Panday A, Elango R, Willis NA (2019) DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20:698–714. https://doi.org/10.1038/s41580-019-0152-0
Sentmanat M, Wang S, Elgin S (2013) Targeting heterochromatin formation to transposable elements in Drosophila: potential roles of the piRNA system. Biochemistry 78:562–571. https://doi.org/10.1134/S0006297913060023
Shang W-H et al (2013) Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev Cell 24:635–648. https://doi.org/10.1016/j.devcel.2013.02.009
Srivastava S, Foltz DR (2018) Posttranslational modifications of CENP-A: marks of distinction. Chromosoma 127:279–290. https://doi.org/10.1007/s00412-018-0665-x
Stadler G et al (2013) Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat Struct Mol Biol 20:671–678. https://doi.org/10.1038/nsmb.2571
Steiner FA, Henikoff S (2015) Diversity in the organization of centromeric chromatin. Curr Opin Genet Dev 31:28–35. https://doi.org/10.1016/j.gde.2015.03.010
Sullivan BA, Karpen GH (2004) Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol 11:1076–1083. https://doi.org/10.1038/nsmb845
Sun FL et al (2004) cis-Acting determinants of heterochromatin formation on Drosophila melanogaster chromosome four. Mol Cell Biol 24:8210–8220. https://doi.org/10.1128/MCB.24.18.8210-8220.2004
Takada M et al (2017) FBW7 Loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res 77:4881–4893. https://doi.org/10.1158/0008-5472.CAN-17-1240
Talbert PB, Henikoff S (2017) Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol 18:115–126. https://doi.org/10.1038/nrm.2016.148
Talbert PB, Henikoff S (2018) Transcribing centromeres: noncoding RNAs and kinetochore assembly. Trends Genet 34:587–599. https://doi.org/10.1016/j.tig.2018.05.001
Tiwari B, Jones AE, Abrams JM (2018) Transposons, p53 and genome security. Trends Genet 34:846–855. https://doi.org/10.1016/j.tig.2018.08.003
Tolstorukov MY, Volfovsky N, Stephens RM, Park PJ (2011) Impact of chromatin structure on sequence variability in the human genome. Nat Struct Mol Biol 18:510–515. https://doi.org/10.1038/nsmb.2012
Topp CN, Zhong CX, Dawe RK (2004) Centromere-encoded RNAs are integral components of the maize kinetochore. Proc Natl Acad Sci USA 101:15986–15991. https://doi.org/10.1073/pnas.0407154101
Tubbs A, Nussenzweig A (2017) Endogenous DNA damage as a source of genomic instability in cancer. Cell 168:644–656. https://doi.org/10.1016/j.cell.2017.01.002
Varvara PV et al (2019) gamma-H2AX: A potential biomarker in breast cancer. Tumour Biol 41:1010428319878536. https://doi.org/10.1177/1010428319878536
Vijg J, Suh Y (2013) Genome instability and aging. Annu Rev Physiol 75:645–668. https://doi.org/10.1146/annurev-physiol-030212-183715
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297:1833–1837. https://doi.org/10.1126/science.1074973
Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, Allshire RC (2003) RNA interference is required for normal centromere function in fission yeast. Chromosome Res 11:137–146
Voullaire LE, Slater HR, Petrovic V, Choo KH (1993) A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: activation of a latent centromere? Am J Hum Genet 52:1153–1163
Wallrath LL, Elgin SC (1995) Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev 9:1263–1277. https://doi.org/10.1101/gad.9.10.1263
Wang B, Zhang Z, Xia S, Jiang M, Wang Y (2019) Expression of gamma-H2AX and patient prognosis in breast cancer cohort. J Cell Biochem 120:12958–12965. https://doi.org/10.1002/jcb.28567
Warburton PE et al (1997) Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr Biol 7:901–904. https://doi.org/10.1016/s0960-9822(06)00382-4
Warnecke T, Batada NN, Hurst LD (2008) The impact of the nucleosome code on protein-coding sequence evolution in yeast. PLoS Genet 4:e1000250–e1000250. https://doi.org/10.1371/journal.pgen.1000250
Washietl S, Machne R, Goldman N (2008) Evolutionary footprints of nucleosome positions in yeast. Trends Genet 24:583–587. https://doi.org/10.1016/j.tig.2008.09.003
Waterborg JH (1990) Sequence analysis of acetylation and methylation in two histone H3 variants of alfalfa. J Biol Chem 265:17157–17161
Weitzman MD, Fradet-Turcotte A (2018) Virus DNA replication and the host DNA damage response. Annu Rev Virol 5:141–164. https://doi.org/10.1146/annurev-virology-092917-043534
Wellinger RJ, Zakian VA (2012) Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics 191:1073–1105. https://doi.org/10.1534/genetics.111.137851
Wendte JM, Pikaard CS (2017) The RNAs of RNA-directed DNA methylation. Biochim Biophys Acta 1860:140–148. https://doi.org/10.1016/j.bbagrm.2016.08.004
Wierman MB, Smith JS (2014) Yeast sirtuins and the regulation of aging. FEMS Yeast Res 14:73–88. https://doi.org/10.1111/1567-1364.12115
Wright WD, Shah SS, Heyer W-D (2018) Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 293:10524–10535. https://doi.org/10.1074/jbc.TM118.000372
Wu RA, Upton HE, Vogan JM, Collins K (2017a) Telomerase mechanism of telomere synthesis. Annu Rev Biochem 86:439–460. https://doi.org/10.1146/annurev-biochem-061516-045019
Wu Z, Liu J, Zhang QD, Lv DK, Wu NF, Zhou JQ (2017b) Rad6-Bre1-mediated H2B ubiquitination regulates telomere replication by promoting telomere-end resection. Nucleic Acids Res 45:3308–3322. https://doi.org/10.1093/nar/gkx101
Xin H, Liu D, Songyang Z (2008) The telosome/shelterin complex and its functions. Genome Biol 9:232. https://doi.org/10.1186/gb-2008-9-9-232
Yamamori T et al (2010) SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res 38:832–845. https://doi.org/10.1093/nar/gkp1039
Zhang W et al (2016) SIRT1 inhibition impairs non-homologous end joining DNA damage repair by increasing Ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 7:13538–13550. https://doi.org/10.18632/oncotarget.6455
Zhou M, Law JA (2015) RNA Pol IV and V in gene silencing: rebel polymerases evolving away from Pol II's rules. Curr Opin Plant Biol 27:154–164. https://doi.org/10.1016/j.pbi.2015.07.005
Acknowledgements
We would like to thank the members of the Riddle lab for helpful discussions. Work in the Riddle laboratory is supported by the National Science Foundation under Grant No. 1552586.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feng, J.X., Riddle, N.C. Epigenetics and genome stability. Mamm Genome 31, 181–195 (2020). https://doi.org/10.1007/s00335-020-09836-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-020-09836-2