
Abstract
Aging is a developmental process that occurs through epigenetic reprogramming that involves nine hallmark characteristics, most notably genomic instability. During physiological development, chromatin is modified, reorganized, and de-compacted in order for DNA to be transcribed, replicated, and repaired. The most prominent histone modifications include acetylation, methylation, ubiquitylation, ADP-ribosylation, phosphorylation, and sumoylation. Younger cells/tissues are characterized by greater global methylation. Global DNA demethylation in aging occurs mainly at repetitive DNA elements and in genome regions with facultative heterochromatin, which leads to overall deheterochromatinization of the genome.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Entering the Puzzling World of Aging
Aging has long been considered as a major socioeconomic problem. It leads to consecutive worsening of the overall organismal health, which in turn results in steady increase of public expenses in medical and social fields (Upadhya et al. 2015; Rolf 2015). The mere fact that the overall life expectancy is steadily increasing and that almost all human populations are characterized by a growing number of elderly people turns this problem into a huge burden (Guerrero et al. 2015; McKenzie et al. 2015). Hence, it is of utmost importance for all governments to assure healthy aging of its elderly people which sounds as an impossible task without full comprehension of the general mechanisms of aging. The last has been a challenge for scientists in the field of gerontology for many years, even for decades. And this is not surprising at all. Aging is a complex biological process which leads to loss of functional reserve of multiple organ systems and to increased susceptibility to stress and age-associated maladies like cancer , neurodegenerative diseases, diabetes, etc. It is also associated with increased incidence of genetic mutations and prevalence of chronic diseases which finally lead to functional dependence and an inadequate life (Campisi 2000; Miquel 2014). Determined by a combination of genetic and environmental factors, the process of getting old is highly individualized and yet poorly understood. Extensive studies are under way with the main emphasis being set on elucidation of the intimate molecular mechanisms of the aging process.
Recently, some researchers in the field of aging (López-Otín et al. 2013) have been able to shortlist the nine most common hallmarks of the aging process and denoted them as major. These nine major hallmarks include genomic instability , telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence , stem cell exhaustion, and altered intercellular communication. Deep understanding of these common aging features could be a major step toward development and application of specific pharmaceutical approaches, the aim of which would be to ameliorate, slow, and successfully manipulate the process of getting old. Of course, scientists in the field are far from getting involved in the search for the elixir for eternal life, but do endeavor to spend lots of efforts in order to get a clearer picture of the most intimate mechanisms of aging. The task could be very hard because these well classified hallmarks of aging seem interdependent making studies in the field even more complicated. For example, DNA damage could lead to genomic instability but could be a consequence of it. Many factors, some extrinsic like UV irradiation, chemicals, and genotoxins, and others, intrinsic like ROS (reactive oxygen species) production, account for DNA damage and are one of the main culprits for the aging of the cells and the organism (Garinis et al. 2008). The picture gets even more complicated since the metabolic stress that all cells undergo could be the primary cause for increased ROS production. Apparently following this line we enter in the vicious circle of trying to understand who is who and what is the cause and what the consequence of aging. No doubt, therefore, that the complexity of aging is considered as “one of the major stumbling blocks of all biological investigations of aging” (Martin 2005).
Bearing in mind that all organisms have DNA and that the proper functioning of the genetic material is crucial for the cellular and organismal homeostasis, it is easy to predict that the genome itself could be the foundation on which aging acts. No doubt that if we scrutinize the way by which the genome ages and moreover, its dynamics during aging, we shall be able to understand the general mechanisms of this complicated process.
2.2 Chromatin as a Modulator of Cellular Fate
2.2.1 The Consecutive Steps of Chromatin Compaction in the Eukaryotic Nucleus
Each of the 60 trillion cells that make up the human body (with some minor exceptions like the human erythrocytes) contains 2 m of genomic DNA in its nucleus. This is quite a huge amount of genetic material that has to be packed and organized in the confined space of the cell nucleus (Fig. 2.1). The organization of DNA and, importantly, its proper functioning recently turned into one of the most interesting puzzles in biology. Two important issues have to be solved by the cells. The first is about the way the genomic DNA is packaged to fit the limited space in the cell nucleus which is only 10 μm in diameter, and the second question is about the way DNA works in this very compacted state. As a matter of fact almost all articles regarding chromatin , epigenetics , and DNA start with one and the same sentence, and it goes like that: “DNA is not naked in the cells but is organized together with histone proteins in chromatin.” Indeed chromatin is the platform where DNA meets histones—the last are considered to be at the heart of DNA compaction, its regulation, and dynamics (Hayes et al. 1990; Littau et al. 1965; Wolffe 1998). Together with it they build the nucleosomes. Each nucleosome consists of two molecules of each of the four core histones (H2A, H2B, H3, and H4), around which 147 bp of DNA are wrapped together. The nucleosome represents the basic repeat unit of chromatin (Richmond et al. 1984; Luger et al. 1997). It is widely accepted that this is the first level of chromatin compaction. By a physical point of view, the nucleosome represents an obstacle to all processes that require access to the molecule of DNA.

A drawing of chromatin compaction in the eukaryotic nucleus. DNA in the eukaryotic nucleus is organized together with histone proteins in chromatin. Nucleosomes are the main building blocks of chromatin which further compact together with linker histones in higher-order chromatin structures thus preserving the genome against stress but also maintaining its functionality
Therefore, chromatin has to be modified, reorganized, and de-compacted in order DNA to be transcribed, replicated, and repaired. This process of chromatin reorganization is done by specialized enzyme complexes, called chromatin modifying and chromatin remodeling complexes, which can chemically modify histones and can slide nucleosomes along the molecule of DNA. Nucleosomes are not only basic units of chromatin compaction but also exert important regulatory functions and manipulate the activity of the genome. They form nucleosome arrays, which in relation to the electron microscopic images have poetically been called the “beads-on-a-string” structure (Thoma and Koller 1977; Rattner et al. 1982). The diameter of this structure is approximately 11 nm and represents the second level of chromatin compaction. Regarding the size of the eukaryotic genome, it is obvious that this level of compaction is quite insufficient for stuffing DNA in the nucleus. It has been further shown that chromatin additionally compacts and forms 30 nm in diameter fibers (Robinson and Rhodes 2006). Such folding of the nucleosome arrays into a compact filament with a diameter of about 30 nm in a salt-dependent manner was reported in numerous in vitro studies (Finch and Klug 1976; Thoma et al. 1979; Widom and Klug 1985; Huynh et al. 2005; Robinson et al. 2006). Evidence for the presence of the 30 nm chromatin fiber in nuclei has also been provided from X-ray diffraction and electron microscopy analyses (Langmore and Schutt 1980; Andersson et al. 1982; Marsden and Laemmli 1979). 30 nm “fiber-like” structures were detected when chromatin fragments were released from human cell nuclei and were also visualized by atomic force microscopy of reconstituted chromatin and of chromatin fragments from yeast nuclei (Georgieva et al. 2012, 2015; Prieto et al. 2012; Gilbert et al. 2004).
However, in contrast to the nucleosome structure, which is relatively well understood, the detailed organization of these 30-nm diameter rod-like structures is considered as quite controversial. Recently, their existence in vivo has been questioned thus opening ardent discussions of how exactly chromatin is structured above the 11-nm chromatin filaments (Fussner et al. 2012; Maeshima et al. 2014). The dispute probably has been induced by the different methodologies exploited for studying of this particular level of chromatin organization. It is true that some of the most meaningful results on this structure were obtained by in vitro experiments (Robinson et al. 2008; Robinson and Rhodes 2006; Routh et al. 2008). Though, for more than four decades, lots of data were obtained by both in vivo and in vitro experiments favoring the existence of the 30 nm chromatin fibers. Interestingly, the obtained results have proposed the existence of at least two different rod-like fibers in the nuclei of eukaryotes: the first with a diameter of 30 nm (the so-called “one-start model”) and the second with a diameter of 24–26 nm (the “two-start model”), both, as noted by Grigoryev and coworkers (2009) able to appear consecutively in a single fiber. Importantly, in a critical review by Ghirlando and Felsenfeld (2013), it was stated that the 30 nm fiber is not an indicator of the transcriptional status of chromatin but rather represents a structurally more compacted state of organization.
2.2.2 The Elusive Higher-Order Chromatin Structures
Chromatin organization above the 10 and 30 nm in diameter structures is believed to employ important regulatory roles on the genetic information. Treatment of isolated nuclei or chromosomes with high salt solutions that remove histones resulted in the formation of a “halo” of supercoiled DNA loops surrounding a denser core (Cook and Brazell 1975; Earnshaw and Laemmli 1983). It has been proven that these chromatin loops were anchored to an insoluble proteinaceous nuclear matrix (or karyoskeleton) by DNA sequences termed as matrix attachment regions (MARs) and scaffold attachment regions (SARs), respectively.
Another aspect of the higher-order structure of chromatin is manifested by its spatial, 3D organization and motion. In the cell nucleus, each chromosome occupies a limited volume, chromosome territory, and seems relatively static being physically anchored in this distinct and limited nuclear space (Figs. 2.1 and 2.2). However, the findings from live microscopy and other experiments that enable tracking of chromatin dynamics in vivo have revealed large-scale chromatin motions and have proved the functional significance of these motions for the cells and the organism (Gasser 2002; Chuang et al. 2006). Three principle types of chromatin motion were recognized and were accepted as general. Interestingly, the three appeared quite different in time scale, range, and frequency of the movements as well as in energy dependence (Soutoglou and Misteli 2007). Moreover, it has been shown that these chromatin spatial rearrangements influence cellular fate and aging of the organisms (Chandra et al. 2015).

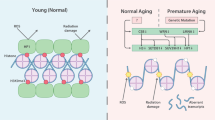
A schematic representation of higher-order chromatin loop organization in three cellular conditions: a normal young cell, an aging cell, and a pathologically changed cell from a patient with Hutchinson–Gilford Progeria syndrome (HGPS). Normally, interphase chromosomes are making numerous contacts with the nuclear lamina. Due to changes in chromatin organization and perturbations in lamina organization during the processes of cellular aging and in premature aging syndromes, many of these contacts between the chromosomes and the lamina are lost (shown with arrows in the figure). This leads to formation of longer chromatin loops, reorganization of chromosome territories, and loss of heterochromatin in the nucleus. In cells from the premature aging syndrome, HGPS, the nuclear lamina is severely dilapidated and therefore most of the chromosome contacts with it are disrupted. The diminished contacts between the lamina and the chromosomes in aged cells and in HGPS cells probably alter gene expression in a similar fashion and therefore aging and progeria syndromes represent similar clinical outcomes
Global chromatin reorganization and changes in nuclear architecture with functional aftereffects in the regulation of gene expression are associated with differentiation , development and aging (Solovei et al. 2009; Oberdoerffer and Sinclair 2007). According to the conventional view of the three-dimensional (3D) genome organization within the nucleus, heterochromatin preferentially localizes at the nuclear periphery, whereas euchromatin preferentially resides in the nuclear interior. A remarkable exception of this conventional nuclear architecture is the inverted chromatin organization in photoreceptor cells of the nocturnal mammalian eye. Using FISH, some authors were able to characterize in detail the higher-order chromatin dynamics during terminal differentiation of rod cells of mouse retinas (Solovei et al. 2009). Interestingly, at birth rod nuclei have a conventional architecture, but postnatal differentiation of rod cells is accompanied with global spatial movement of higher-order chromatin structures and with the establishment of the so-called “inverted” nuclear organization.
Finally, in terminally differentiated mouse rods heterochromatin concentrated in the nuclear center, whereas euchromatin as well as nascent transcripts and splicing machinery lined the nuclear border (Solovei et al. 2009). The functional effect of this inverted pattern of nuclear architecture is to provide additional sensitivity of nocturnal mammal eye in the poor light environments. These results unambiguously prove that chromatin dynamics exert functional significance during organismal lifespan .
2.3 Chromatin: Static and Dynamic at the Same Time
Cells are persistently exposed to intrinsic as well as extrinsic environmental signals that could destine cellular fate toward proliferation, differentiation , quiescence, senescence , or malignancy. All these processes are accompanied and governed by dynamic reorganization of chromatin (Shin et al. 2011; Apostolou and Hochedlinger 2013; Pegoraro and Misteli 2009; Adams 2007; Misteli 2010; Burton and Torres-Padilla 2014). We have tried to demonstrate some of the best known chromatin large-scale reorganization during normal development , aging, and premature aging syndromes in Fig. 2.2. In order to regulate all biological processes related to the molecule of DNA, chromatin has to hold a flexible, versatile structure that can respond to internal and external signals during cellular lifespan .
To answer these challenges, chromatin exhibits a highly dynamic equilibrium between an open conformation (e.g., 11-nm beads-on-a-string) up to much more compacted, regarded even as a closed chromatin state (the so-called higher-order structures). This dynamics assures both accurate expression of the genetic information as well as promises genomic stability. For example, in the time course of the cell cycle, from mitosis through interphase up to the next mitosis, chromatin undergoes global structural reorganization—from fully condensed mitotic chromosomes through less condensed, more diffused interphase chromatin conformation and then again to compacted chromosome entities. In order to meet the requirements of DNA-related processes, chromatin is not expected to be steadily and uniformly compacted across the genome in vivo , but to be dynamically modulated by local differences in chromatin components including numerous architectural and regulatory proteins as well as complex combinations (cross-talks) among diverse set of epigenetic modifications. No doubt that the regulation of chromatin dynamics comes through a variety of mechanisms including DNA methylation (Karymov et al. 2001; Ma et al. 2005), posttranslational modifications (PTMs) of histones (Ito 2007; Gelato and Fischle 2008), histone variants (Happel and Doenecke 2009; Clausell et al. 2009), recruitment of chromatin remodeling complexes (Corona et al. 2007; Clapier and Cairns 2009), and noncoding RNAs (Li 2014; Dhanasekaran et al. 2013). All these epigenetic regulators act in concert to exert the complex mechanisms that modulate chromatin conformation which in turn regulates gene expression in development , differentiation , aging, and disease. For example, these epigenetic marks as well as the level of chromatin condensation are distinct between normally proliferating, cancerous, and senescent cells (Oh et al. 2013; Rodriguez-Paredes and Esteller 2011).
Due to the development of new techniques, chromatin dynamics has been under extensive studies recently. It is reasonable to believe that the versatile elaboration of experimental techniques such as FISH (fluorescent in situ hybridization), 3C (chromosome conformation capture), and its high-throughput modifications (4C, 5C, Hi-C, ChIA-PET, etc.) (Dekker et al. 2002; Sanyal et al. 2011), together with AFM (atomic force microscopy), ChCA (chromatin comet assay) (Georgieva et al. 2008, 2012), and the very recently developed high resolution live microscopy, and the gathering of valuable information about higher-order chromatin structure and its dynamics will exponentially increase in the future.
2.4 Chromatin Structure and Dynamics as Major Players in Aging
Chromatin has been accepted as a major player in aging as it regulates the plasticity of the genome and governs its susceptibility to intrinsic and extrinsic DNA damaging signals. It also modulates the contacts of DNA with all factors that participate in replication, transcription , and repair. The proper contacts among DNA and all these factors guarantee their accurate functioning, contribute to the preservation of the genome against genotoxic stress , and prevent the accumulation of mutations which could be detrimental for the cells (Downs and Cote 2005; Cho et al. 2004; Seeber et al. 2014; Rodrigues et al. 2014).
As we have already discussed, the nature of chromatin is very contradictory. At one hand, it is regarded as a static unit that guards the genome and protects the genetic material against DNA damage. On the other hand, chromatin appears quite dynamic. It “breaths in” and “out” with the surrounding environment and thus allows the cells to adapt by changing the expression of certain genes. In some cases, this dynamic property appears to counter the aging process and to extend organismal lifespan (Longo and Kennedy 2006; Gotta et al. 1997; Kennedy et al. 1997). However, the stochastic component of chromatin structure might also contribute to the breakdown of nuclear, cellular, and tissue functions and consequently to lead to aging and age-associated diseases (Lazarus et al. 2013; Sedivy et al. 2008). Generally, aging involves major changes in chromatin structure and organization (Zane et al. 2014; Pegoraro and Misteli 2009; Muñoz-Najar and Sedivy 2011). It has long been believed that during aging, nuclei show loss of heterochromatin (Ishimi et al. 1987). Global changes in chromatin organization have been found to be associated with aging including the formation of senescence -associated heterochromatin foci (SAHF) in euchromatic DNA and a gradual loss of perinuclear heterochromatin (Oberdoerffer and Sinclair 2007). Deterioration of chromatin organization triggers in turn transcriptional alterations, accumulation of DNA damage, and increased genomic instability (Pegoraro and Misteli 2009; Feser and Tyler 2011; O’Sullivan and Karlseder 2012).
Though recent studies demonstrate that the situation is much more complicated as heterochromatin can be either reduced or increased during senescence (Sedivy et al. 2008; Pegoraro and Misteli 2009). It has been speculated that these SAHF silence proliferation-promoting genes and thus lead to senescence-associated phenotypes (Corpet and Stucki 2014). Remarkably, although SAHF appear to result from the condensation of almost entire chromosomes, DNA sequences that are typically contained in constitutive heterochromatin, such as pericentromeres and telomeres, actually appear to be excluded from the bulk of the condensed chromosomes (Zhang et al. 2009a; Narita et al. 2003). This was further proven by other authors who have shown that the shortened telomeres in mice lacking telomerase have reduced heterochromatin compared to telomeres from normal cells (Benetti et al. 2007a). The molecular mechanism of SAHF formation remains poorly understood although histone chaperones Asf1a and HIRA have been reported to play a role in their formation (Zhang et al. 2005). Other authors have even suggested that SAHF are a novel type of chromatin condensation involving alterations in the way by which DNA interacts with its binding proteins (Funayama et al. 2006).
They have suggested that the induction of a specific senescence phenotype is accompanied by loss of linker histone H1 from chromatin and accumulation of HMGA2 at its place. This boldly highlights the significance of chromatin structural proteins such as H1 and HMGA2 for the ongoing process of chromatin structure remodeling during aging.
It can be inferred by this that the eu- and heterochromatic states of chromatin are quite dynamic during organismal lifespan , and it is quite easy to envision that this dynamics is basically directed by epigenetic mechanisms.
2.5 Epigenetic Changes in Chromatin During Aging
2.5.1 DNA Methylation : The Aging Clock of the Organism
Epigenetics plays crucial role during development . It shapes the way the genome works in response to all kinds of changes in the surrounding environment (Wolffe and Matzke 1999; Wolffe 1998). Recent twin studies and studies with long-lived families have shown that only 20–30 % of the variations in the aging among different individuals are determined by genetic factors. Interestingly, these are relevant mainly for survival at advanced age. The other 70–80 % of variations have been shown to be associated with stochastic events, i.e., certainly to some nongenetic factors (Herskind et al. 1996). Essential for development, epigenetics inevitably becomes misregulated and misdirected during disease and aging, and importantly, these changes are more or less conserved among all species (Sedivy et al. 2008; Wood and Helfand 2013; Zane et al. 2014).
A major part of the epigenetic research has been focused on the study of modifications of DNA and histone proteins, as these are the mechanisms that generally influence chromatin structure and organization. DNA methylation , which is the first epigenetic phenomenon that has been linked to aging and disease, is also the best-studied DNA modification (Riggs 1975; Iyer et al. 2011; Fraga et al. 2005a).
Recently, some interesting dynamics of DNA methylation during aging have been explored (Zane et al. 2014; Muñoz-Najar and Sedivy 2011; Lazarus et al. 2013). It has been demonstrated that there is a consistent reduction in global DNA methylation during aging (Singhal et al. 1987; Wilson et al. 1987) while at certain places, namely the CpG islands of gene promoters, DNA methylation is increased with age (Kim et al. 2005). In general, global DNA demethylation in aging occurs mainly at repetitive DNA elements and in genomic regions with facultative heterochromatin which leads to overall deheterochromatinization of the genome (Sedivy et al. 2008). In contrast, hypermethylation of CGIs (CpG islands) is found mainly at promoters of stem cells’ genes and also at promoters of tumor suppressor genes (Issa et al. 2001; Waki et al. 2003). Both epigenetic phenomena explain two of the hallmarks of aging (López-Otín et al. 2013): the observed exhaustion of stem cells during aging and the higher incidence of cancer with the advancement of age. The first is due to increased methylation at the promoters of stem cells genes, especially those responsible for their pluripotency, while the second results from the aberrant hypermethylation of promoters of tumor suppressor genes. Interestingly, it has been observed that there is an increase in 5-methylcytosines within the ribosomal DNA (rDNA) clusters in livers of old rats, which could explain the decrease in ribosomal RNA (rRNA) levels that also occurs during aging (Oakes et al. 2003). And though DNA methylation patterns at first sight seem quite contradictory, there is logic for exploiting it as an aging marker.
Not surprisingly, the level of DNA methylation has been proposed as a biological clock for aging. For example, some authors (Mitteldorf 2015; Hannum et al. 2013) offer the level of DNA methylation in stem cell niches in an aging organism as a marker, even as a biological clock for assessing the age of the organism. In a fascinating work by Horvath (Horvath 2013), a multi-tissue predictor of age that allows estimation of DNA methylation age of most human tissues has been made freely available to the public and is gaining lots of popularity (Marioni et al. 2015; Horvath et al. 2015).
Advanced age is always associated with increased risk for cancer . On the other hand, epigenetic alterations are hallmarks of both aging and cancer. Therefore, it is normal to assume that aging-associated DNA methylation changes actively contribute to cancer susceptibility and growth (Fraga et al. 2007). No doubt that both processes (aging and cancer) share almost the same DNA methylation marks. Table 2.1 summarizes the plethora of data concerning the role of DNA methylation in aging, cancer, and premature aging syndromes. What catches the attention immediately is the fact that most DNA patterns are shared among different cases. For example, global DNA hypomethylation of repeat-rich regions is observed in numerous cancers (Ehrlich 2009). DNA hypomethylation of repeat elements has been proposed to contribute to cellular transformation by promoting chromosomal rearrangements and elevating mutational rate. Apart from hypomethylation of repeat elements, loss of methylation from genic regions also causes aberrant gene expression and promotes tumorigenesis (Zane et al. 2014). In addition, promoters of genes that are targets of hypermethylation during aging overlap with DNA hypermethylated genes in many cancers (McGarvey et al. 2008). For example, aberrant hypermethylation at promoters of CDKN2A, LOX, RUNX3, and TIG1, which act as tumor suppressor genes, has been detected during aging (Yanagawa et al. 2007; So et al. 2006a, b) and was further accepted as a strong evidence that there is age-dependent onset of different types of cancer.
2.5.2 The Epigenetic Make-Up of Histone Proteins Serves as a Strong Regulator of Genome Activity
The epigenetic characteristics of young and old cells are not limited only to DNA methylation . Histones (core histones H2A, H2B, H3 H4, and the linker histones, H1) can undergo diverse and reversible posttranslational modifications that occur predominantly on their unstructured “tail” domains. Currently, more than ten different types of histone modifications are known to affect chromatin structure and function. The most prominent histone modifications include acetylation, methylation, ubiquitylation, ADP-ribosylation, phosphorylation, sumoylation, and the list is ever growing (Bannister and Kouzarides 2011; Zhang et al. 2009a).
These covalent marks can be written and erased by site-specific enzymes such as histone acetyltransferases (HATs) and deacetylases (HDACs), histone methyltransferases (HMTs) and demethylases (KDM) (Cuthbert et al. 2004; Shi et al. 2004; Wang et al. 2004), ubiquitin ligases and deubiquitinases, and many others specific for a particular modification. The known histone modifications and some of their functional consequences are very well discussed in (Bannister and Kouzarides 2011). The best known of these PTM’s are shortlisted in Table 2.2, where their role in aging, cancer , and the premature aging syndromes are very well discussed. Posttranslational modifications not only alter the interaction of histone proteins with DNA but also influence inter-nucleosomal interactions and thus affect the overall chromatin structure. Moreover, these modifications could also recruit specific chromatin-associated proteins, like HP1 (heterochromatin protein 1), which recognizes and binds to H3K9me or the polycomb-group proteins (PcG), which recognize and bind to H3K27me or other complexes necessary for remodeling and maintenance of the global chromatin higher-order organization. These are also important for the establishment of distinct, probably specific, chromatin structures in defined genomic regions. Thus, local series of less compact, a more active open chromatin regions and a more condensed, closed chromatin patches were observed along the chromatin fiber (Dekker 2008; Filion et al. 2010; Gilbert et al. 2004). There are many studies showing the significance of PTM’s of histones for the aging process. Moreover, many of them have been accepted as markers of aging, cancer, and HGPS.
2.5.2.1 Saccharomyces cerevisiae: The Golden Model for Chromatin and Aging Research
Early work on epigenetics , chromatin , and aging has been extensively done on Saccharomyces cerevisiae. In yeast, aging can be studied using two main approaches: replicative lifespan (RLS) and chronological lifespan (CLS). RLS is defined as the number of buds produced before cell death. In practice, the replicative lifespan is measured by counting the number of divisions achieved by a cell whose buds are removed one by one by microdissection.
Yeast replicative aging is comparable to aging phenomena observed in asymmetrically dividing cells of higher eukaryotes such as stem cells (Henderson and Hughes 2014; Lindstrom et al. 2011; Lindstrom and Gottschling 2009). Alternatively, the yeast chronological aging is akin to the aging of nondividing cells such as neurons (Longo and Fabrizio 2012; Longo and Kennedy 2006). CLS is defined as the time a cell survives in a nondividing state, with survival being measured as cell wall integrity or as ability to form a colony. Aging is then characterized based on the cells distribution in regard to their chronological lifespans, obtained by measuring survival with time in a stationary phase culture. Both models of yeast aging are accepted as golden models helping to understand aging in higher eukaryotes.
In these cells, inactivation of the histone deacetylase , Sir2, has led to shortened replicative lifespan (Kennedy et al. 1997). Conversely, activation of Sir2 extended lifespan. This phenomenon could be well apprehended if the logic behind the molecular mechanisms is explained in detail. The anti-aging effect of Sir2 in yeast was, at least in part, due to the translocation of a Sir2-containing protein complex from telomeres to ribosomal DNA (rDNA) repeats. These repeats are prone to recombination and form extrachromosomal rDNA circles (ERCs), which by complicated mechanisms shorten yeast lifespan. At the rDNA repeats, Sir2-mediated histone deacetylation, and consequent heterochromatinization by the Sir2-associated proteins (Sir3 and Sir4), prevents recombination and formation of ERCs, thereby extending lifespan (Kaeberlein et al. 2007; Kennedy et al. 1997). Notably, orthologs of Sir2 have been shown to possess strong anti-aging functions in many other species, including nematodes, flies, and human (Herskovits and Guarente 2014; Guarente 2013). In particular, transgenic overexpression of mammalian SIRT1 (the closest homolog to invertebrate Sir2) improves aspects of health during aging but does not increase longevity in mice (Herranz and Serrano 2010). The mechanisms of the beneficial effects of SIRT1 are complex and interconnected, including improved genomic stability that prevents the genome against fast aging.
Another epigenetic mark that turned out to be firmly connected to aging is the acetylation of histones. Histone acetylation and deacetylation seem quite important epigenetic modifications throughout the whole organismal lifespan . For example, increased global levels of H4K16ac were detected in old cells. Higher levels of H4K16ac have been demonstrated also at specific regions of the genome of old cells, for example, at the X core and X elements within the telomeric regions (Dang et al. 2009). A correlation between the increased levels of H4K16ac with age and a decreased silencing of reporter genes inserted at these telomere proximal DNA elements was also reported in this chapter (Dang et al. 2009). It has been discussed that the increased levels of H4K16ac in old cells lead to a more open chromatin structure. The N-terminal tail of H4 is a crucial player in the formation of a fully compacted 30 nm chromatin fiber (Dorigo et al. 2003; Robinson et al. 2008). Therefore, it is not surprising that increased H4K16ac is accepted determinative for the more open state of chromatin and for the genome instability observed in aged cells. Interestingly, this specific epigenetic modification has been linked to the linker histone H1, one of the factors responsible for genome stability (Downs and Cote 2005; Downs et al. 2003; Georgieva et al. 2012). It was shown recently that loss of the tumour suppressor protein phosphatase PTEN leads to dissociation of histone H1 from chromatin, elevation of histone H4 acetylation at lysine 16, and decompaction of chromatin (Chen et al. 2014b).
2.5.2.2 Linker Histones: Multiple Roles in Aging and Development
Linker histones are important structural proteins of chromatin . They bind to DNA at its nucleosome entry/exit sites and are involved in the formation and maintenance of higher-order chromatin structure. Studies on H1 mobility in different cell types exposed to various treatments have suggested that modulation of H1-chromatin interactions are one of the earliest events leading to changes in the structure of the chromatin fiber (Catez et al. 2006). Represented by 11 subtypes in higher eukaryotes which are cell and tissue-specific, it is not surprising that linker histones have long been a challenge for scientists. Importantly, it is proven that these histones are essential for development . The reduction of H1 content with 50 % led to severe embryonic defects and finally to death in mice (Fan et al. 2005). Moreover, recently it has been shown that the levels of H1, especially of H1c (one of the subtypes of H1 family), are crucial for the process of terminal differentiation of retina rod cells which requires global reorganization of chromatin and concentration of heterochromatin in the nuclear center (Popova et al. 2013). As already has been discussed in the previous sections, the linker histone H1 is linked to aging. Its loss during senescence is associated with the formation of SAHF (Funayama et al. 2006). As was mentioned, studies on linker histones in higher eukaryotes are impeded by the fact that higher eukaryotes have many subtypes of H1 and some of them are replaceable, i.e., they can be exchanged by other subtypes of linker histones. We have summarized the best known changes in linker and core histones together with other structural chromatin proteins in Table 2.3 during aging, cancer , and HGPS. What is easily seen and very well expressed is the link among these changes in the three discussed states of the organism: the process of getting old, the process of malignization, and the detrimental premature aging features of HGPS. Knowing these changes is a prerequisite for all steps toward successful managing of them. For this, we need model organisms that could offer the most relevant platform for these studies.
Because of its single-copy gene coding for H1, which furthermore is nonessential, the yeast S. cerevisiae is a wonderful model for studies of linker histones. S. cerevisiae cells offer vivid opportunities for studying the role of linker histones in higher-order chromatin organization in vivo and, moreover, the role of this structure in aging. Recently, we have demonstrated that the yeast linker histone , Hho1p, is involved in the regulation of the aging processes. Mutant yeast cells lacking the gene for the linker histone are viable but inherit highly disordered higher-order chromatin structures which result in perturbed chronological lifespan (Uzunova et al. 2013).
2.5.3 The Elusive Higher-Order Chromatin Structures Dynamics during Aging
During the past decade, scientists have started to see some of the principles of chromatin folding dynamics and their implication in different cellular processes. The first discovery was that individual chromosomes occupy discrete domains in the interphase nucleus—named chromosome territories (Tark-Dame et al. 2011; Cremer and Cremer 2006). This finding raised a lot of discussions in the field of chromatin biology and induced further investigations which led to the observation that the mammalian interphase chromosomes are made up of a large number of structural loops, each of which is on average ~1 Mb, that correspond to DNA replication units (Ryba et al. 2010). Generally, chromatin loops are accepted as an important aspect of chromatin organization. They represent the so-called higher-order chromatin structures and demonstrate high dynamics during aging and development . They bring together distant regulatory elements that control gene expression, such as promoters and enhancers (Kadauke and Blobel 2009), and thus control cellular destiny. For instance, enhancers, insulators, and locus control regions can lie at distances from several kb up to one Mb or more away from the genes they regulate and may be located in cis (upstream or downstream) or in trans (on a different chromosome) (Woodcock 2006).
Thus, chromatin looping is crucial for transcriptional regulation: activation and repression, coordination of initiation and termination, and boundary function. Loop formation is most likely to be a prerequisite for rather than a consequence of transcriptional activation as it occurs prior to gene activation (Palstra 2009). Furthermore, chromatin segments containing active genes loop out from their chromosome territories to reach out a transcription factory for their coordinated expression (Fraser 2006). Figure 2.2 brilliantly illustrates the dynamics in chromatin loop organization during normal development , aging, and some premature aging syndromes. The notion that chromatin loops are important for the overall genome organization is also supported by recent studies in yeast showing that the linker histone is important for the maintenance of bulk chromatin loop organization (Georgieva et al. 2012).
The overall organization of chromatin in loops has been assessed by the method of Chromatin Comet Assay (ChCA) which allows estimation of chromatin loop organization at the level of a single cell (Georgieva et al. 2008, 2012). It has been shown that the general loop size of wild type nuclei is in the range of 0.3 Mb, while when the gene for the linker histone has been deleted, the global chromatin decompaction appeared and the increase in the size of chromatin loops reached 0.4 Mb. This proves that the yeast linker histone, Hho1p, is involved in the global chromatin compaction, loop formation, and probably in the organization of loop attachment sites. Interestingly, this compaction proved to be also important for the normal aging of these cells as further experiments in following the chronological lifespan of the mutants have demonstrated that the absence of the linker histone leads to features resembling premature aging phenotypes (Uzunova et al. 2013), thus strengthening the idea that chromatin higher-order organization is a prerequisite for cellular normality.
In conclusion, chromatin loops can exert different but very important functions in the nucleus. At one hand, they could bring distantly located sequence elements into spatial proximity, thus allowing communication between these sites and proper transcription or repair. It could also induce the opposite, spatial segregation of certain genomic regions from each other ensuring their independent functions. On the other hand, bulk chromatin loop organization has an impact on cellular morphology, which links certain chromatin structural units with proper execution of the main cellular processes. The observed participation of chromatin loop organization in the aging control is an intriguing phenomenon which needs additional investigations in order to reveal the exact molecular mechanisms involved in it. The last statement highlights the necessity of further studies on the dynamics of folded chromatin conformations beyond the mere detection of long-range interactions.
2.6 Premature Aging Syndromes and Age-Associated Diseases in the Context of Chromatin
As we have already discussed in the previous sections, many recent works suggest that aging is driven by epigenetic changes and that epigenetic perturbations can lead to different progeroid syndromes. We believe that collectively these works propose that understanding and manipulating the epigenome are a good perspective for improving age-related pathologies and for extending healthy lifespan .
2.6.1 Chromatin Structure, Aging, and the Premature Aging Phenotype of HGPS
Accumulation of comprehensive experimental data endorses the concept of the causal role of the disturbed chromatin structure and function in the process of aging (Pegoraro and Misteli 2009; Misteli 2010; Feser and Tyler 2011; O’Sullivan and Karlseder 2012).
A unique opportunity to better understand the link between chromatin structure and aging offers the premature aging syndrome, called Hutchison–Gilford progeria syndrome (HGPS). HGPS is a rare, sporadic genetic disease, which occurs in approximately 1 in 4 million live births (Pollex and Hegele 2004; Gordon et al. 2014). The average life expectancy of HGPS patients is 12–15 years. During this reduced lifespan they progressively accumulate features that resemble those of an aged individual like severe growth retardation, lipodystrophy, skeletal dysplasia, joint contractures, alopecia, skin and nail defects, progressive cardiovascular disease, heart attacks, and strokes (Merideth et al. 2008).
HGPS is caused by an autosomal dominant mutation in the LMNA gene coding for two proteins, lamin A/C (expressed by alternative splicing), major structural components of the nuclear lamina. Nuclear lamina has crucial role in a plethora of cellular events, in particular it maintains nuclear shape and provides sites for peripheral heterochromatin binding (Goldman et al. 2002). Being key structural proteins of the nucleus, lamins play important roles in epigenetics , chromatin organization, DNA replication, regulation of gene expression, and DNA repair, cell proliferation, and differentiation as well as in viral infections (Dechat et al. 2008; Goldman et al. 2002). At present, over 100 mutations in LMNA gene has been reported causing more than 15 distinct diseases designated as laminopathies (Hegele 2005).
The point mutation C1824T in exon 11 of the LMNA gene leads to the activation of a cryptic splice donor site and to production of an internally truncated form of lamin A, which lacks 50 amino acid residues near the C-terminus. This dominant negative isoform of lamin A is referred to as progerin (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003). In HGPS cells, expression of progerin interferes with lamin A that in turn abrogates nuclear lamina functionality and brings numerous phenotypes, including epigenetic alterations, disorganization of chromatin , abnormal nuclear morphology (Scaffidi and Misteli 2006; Gordon et al. 2014; Kudlow et al. 2007; Dechat et al. 2008; Schreiber and Kennedy 2013). Some of these epigenetic alterations are shortlisted in Tables 2.1, 2.2, and 2.3, where one can find interesting data about DNA methylation, PTMs of histones, and the level of expression of histone proteins in aging, cancer , and HGPS.
Interestingly, most of these epigenetic characteristics are shared among these organismal states and propose good models for studying of age-associated alterations in chromatin and epigenetics . In addition, increased levels of unrepaired DNA damage, genome instability and telomere shortening, altered gene expression, and defective stem cell homeostasis were detected during HGPS development (Scaffidi and Misteli 2006; Kudlow et al. 2007; Dechat et al. 2008; Schreiber and Kennedy 2013). Cells from aged individuals and HGPS patients display intriguing similarities with respect to changes in the epigenome and chromatin organization though in progeroid cells these chromatin changes seem to be more pronounced (Sedivy et al. 2008; Dechat et al. 2008). These include loss of heterochromatin structure, alterations in the patterns of heterochromatin-associated histone PTMs (elevated histone acetylation; decrease of H3K9me3 and H3K27me3; and upregulation of H4K20me3), histone variants (increase of phospho-H2AX and HGPS-specific loss of the centromeric protein CENP-A), and the level of key architectural chromatin proteins (e.g., reduction of heterochromatin protein HP1, chromatin chaperons RBBP4/7, the histone mehtyltransferase EZH2), as well as deregulation of chromatin remodeling complexes (NuRD, the nucleosome remodeling and histone deacetylase complex) (Goldman et al. 2004; Scaffidi and Misteli 2006; Shumaker et al. 2006; Pegoraro and Misteli 2009; Pegoraro et al. 2009).
Generally, in the eukaryotic cell nucleus, heterochromatin is highly organized and may be visualized by electron microscopy as nucleoplasmic heterochromatic foci or peripheral heterochromatin, associated with regions of the lamina (Dechat et al. 2008). Both heterochromatin structures are absent in skin fibroblasts from HGPS patients, pointing out the general loss of heterochromatin in HGPS nuclei (Goldman et al. 2004; Columbaro et al. 2005; Dechat et al. 2008). To highlight the molecular mechanisms involved in the disruption of heterochromatin structures, several research groups examined the level of epigenetic marks—histone PTMs associated with either facultative (H3K27me3) or constitutive heterochromatin (H3K9me3 and H4K20me3) (Goldman et al. 2004; Scaffidi and Misteli 2006; Shumaker et al. 2006; Pegoraro and Misteli 2009).
In addition to changes in histone PTMs, alterations in the amount of some histone variants and key chromatin proteins have been detected in HGPS cells (Tables 2.2 and 2.3). These include increase in γ-H2AX (Scaffidi and Misteli 2006) and loss of the centromeric protein CENP-A (Pegoraro et al. 2009) in HGPS. In contrast, no decrease in CENP-A was detected in physiologically aged cells. Several other nuclear proteins were down-regulated in HGPS including all isoforms of the heterochromatin protein HP1, methyltransferase EZH2, histone deacetylase HDAC1, LAP2 group of lamin A-associated proteins, histone chaperons RBBP4 and RBBP7 (Pegoraro et al. 2009; Scaffidi and Misteli 2006; Shumaker et al. 2006). The reduction of key heterochromatin proteins correlates with the loss of peripheral heterochromatin and abrogation of the normal nuclear architecture.
In human somatic female cells, the inactivated X chromosome can be identified as a heterochromatin domain usually associated with the nuclear lamina. Silencing of inactivated X is provided by H3K27me3 and Xist RNA (Kohlmaier et al. 2004). EZH2 (enhancer of zeste homolog) methyltransferase is a subunit of the Polycomb Repressive Complex 2 (PRC2) that has an essential role in the epigenetic maintenance of repressive chromatin states through trimethylation of H3K27 (Margueron et al. 2009). In contrast, in fibroblast cells from a female HGPS patient, the H3K27me3 mark is lost on the inactive X chromosome with a concomitant downregulation of EZH2 which leads to some decondensation of this chromosome (Shumaker et al. 2006). Importantly, the alterations in chromatin regulation precede the nuclear shape changes (Shumaker et al. 2006) pointing out the pivotal role of chromatin in pathological premature aging.
Furthermore, genome-wide analysis of H3K27me3 distribution revealed patches of decreased H3K27me3 in HGPS nuclei. In particular, gene-poor regions of HGPS fibroblasts experience a reduction of H3K27me3 compared with the control (McCord et al. 2013). As in previous reports, loss of H3K27me3 in HGPS was associated with downregulation of EZH2 since the mRNA level of EZH2 was significantly reduced in four HGPS fibroblast cell lines compared to normal control samples (McCord et al. 2013; Shumaker et al. 2006; Margueron et al. 2009). Genome-wide analyses revealed a correlation between alterations in patterns of H3K27me3 deposition, genome organization, and DNA-lamin A/C interactions in HGPS skin fibroblasts (McCord et al. 2013). Based on this, authors proposed a model in which accumulation of progerin in the nuclear lamina leads to reduction in the repressive histone mark H3K27me3 in heterochromatin (most possibly through the downregulation of EZH2) and disrupts interactions between heterochromatin and nuclear lamina. These changes may then result in transcriptional misregulation and global loss of spatial chromatin compartmentalization (McCord et al. 2013). In addition, downregulation of the constitutive heterochromatin mark H3K9me3 and loss of pericentric heterochromatin were detected in HGPS cells (Scaffidi and Misteli 2006; Shumaker et al. 2006; Pegoraro and Misteli 2009). The loss of heterochromatinization of pericentric regions induces transcription of pericentromeric satellite III repeats (Shumaker et al. 2006).
In contrast to the reduction in H3K9 and H3K27 trimethylation state, an increase of H4K20me3 was detected in progeroid cells (Shumaker et al. 2006), and this may relate to the telomere shortening observed in these cells. Several lines of evidence argue that the disruption of heterochromatin structure in progeria syndrome can be attributed to deregulation of the Nucleosome Remodeling and Deacetylation (NuRD) complex (Pegoraro et al. 2009). NuRD is a ubiquitous ATP -ase-dependent chromatin remodeling complex that has been shown to associate with pericentromeric heterochromatin and to maintain transcriptional repression at specific promoters (Bowen et al. 2004; Helbling Chadwick et al. 2009). NuRD is a multicomponent complex, containing the histone deacetylases HDAC1 and HDAC2, histone-binding proteins RBBP4 and RBBP7, metastasis-associated protein MTA3, the ATPases CHD3 and CHD4 (chromodomain-helicase-DNA-binding protein) as subunits (Zhang et al. 1999). Besides NuRD, histone chaperone proteins RBBP4 and RBBP7 are shared components of Polycomb PRC2 complex involved in the establishment of heterochromatin (Kuzmichev et al. 2002), and RBBP4 is also a subunit of the CAF-1 complex, which assembles chromatin upon DNA replication and DNA damage repair (Verreault et al. 1996).
RBBP4/7 interact with the amino acid residues 562–664 fragment of lamin A in vivo and neither protein binds to progerin, which lacks residues 607–657 of mature lamin A (Pegoraro et al. 2009) demonstrating that RBBP4/7 mediate physical tethering of NuRD to nuclear lamina. Therefore, the accumulation of progerin in HGPS cells abolishes this interaction that consequently affects the proper chromatin remodeling which in turn triggers deregulation of cellular functions. Further, protein levels of several NuRD components including MTA3, RBBP4, and RBBP7 as well as the HDAC1 amount and activity are reduced in HGPS cells and normal cells expressing exogenous progerin (Pegoraro et al. 2009). The observation that shRNA-mediated silencing of HDAC1, MTA3, CHD3, or CHD4 in normal cells was sufficient to recapitulate age-dependent chromatin defects, e.g., loss of H3K9me3 and HP1γ heterochromatin foci, heterochromatin deregulation, and increased DNA damage, indicates the importance of NuRD in aging-associated chromatin defects (Pegoraro et al. 2009). Moreover, the loss of RBBP4, RBBP7, and HDAC1 subunits of NuRD is not limited to premature aging, but reduced levels of these proteins are also a feature of normally aged cells (Pegoraro et al. 2009).
These findings show that the NuRD complex is deregulated in premature as well as normally aged cells and outline the molecular bases of aging-associated chromatin disorganization. Telomere attrition is one out of the numerous hallmarks of aging in mammals (Cao et al. 2011; López-Otín et al. 2013). Accelerated telomere shortening has also been observed in fibroblasts from HGPS individuals (Huang et al. 2008; Decker et al. 2009). Notably, ectopic expression of progerin in normal fibroblasts recapitulates the faster telomere shortening (Decker et al. 2009). Normally, telomeres and subtelomeric regions are enriched in the H3K9me3 and H4K20me3, epigenetic marks that are characteristic of constitutive heterochromatin (Blasco 2007), and the decrease in H4K20me3 has been associated with telomere elongation (Benetti et al. 2007b). Changes in these histone modifications, namely decrease in H3K9me3 and the increase in H4K20me3 (Shumaker et al. 2006) could account for the enhanced telomere attrition in HGPS cells.
The increased level of persistent DNA damage is another typical feature of physiological and premature aging as already discussed. Cells from HGPS patients and from old individuals displayed focal accumulation of phosphorylated histone H2AX (γ-H2AX). The presence of foci containing γ-H2AX is indicative for unrepaired DNA damage which in turn compromises genomic stability (Liu et al. 2005). However, HGPS individuals do not show higher predisposition to neoplasia that is in contrast to the aged individuals and those affected by other premature syndromes in which the risk of cancer development is significantly high (Hennekam 2006; Fraga et al. 2007).
In addition, similar to cells from HGPS patients and aged individuals, the percentage of cells containing multiple prominent phospho-H2AX foci increases in cells depleted of RBBP4 and RBBP7 (Pegoraro et al. 2009) that implicates chromatin remodeling complexes in the induction of chromatin defects associated with premature aging. Therefore, it is worth noting that the occurrence of the observed structural chromatin defects came about prior to DNA damage (Pegoraro et al. 2009). Revealing which of the age-related changes to chromatin causing aging will benefit development of therapeutic strategies for HGPS and other age-related and accelerated aging diseases.
2.6.2 Age-Associated Diseases: A Challenge for the Biology of Aging
Alzheimer’s disease (AD ) is a neurodegenerative disorder, which results from not only genetic but also from environmental factors. Epigenetic mechanisms, such as DNA methylation , chromatin remodeling and miRNAs, which may induce alterations in gene expression, are thought to be involved in Alzheimer’s disease. The most characteristic feature of AD is cognitive impairment that together with neuropathological changes, extracellular amyloid plaques, intracellular neurofibrillary tangles and loss of synapses and neurons are similar in the early-onset and late-onset forms of the disease. Both forms the early- and late-onset of AD have genetic components. Mutations in three genes identified in the early-onset familial form, i.e., amyloid precursor protein (APP), presenilin-1, and presenilin-2, established the central role of amyloid in the disease. However, it should be noted that mutations in these genes are present in only 13 % of patients with the early-onset form of the disease. The technological advances, such as large-scale genome-wide association studies, have led to identification of more than ten risk genes for the late-onset form of AD (Bettens et al. 2013).
Early onset familial AD accounts for less than 1 % of all AD cases (Lambert and Amouyel 2007). Early-onset Alzheimer’s appears to be linked to a genetic defect on chromosome 14, to which late-onset Alzheimer’s is not linked (Campion et al. 1999). The genes implicated in these forms of the disease are the gene encoding for APP, located on chromosome 21q21, the gene encoding for presenilin 1 (PSEN1), located on chromosome 14q24.3, and that encoding for presenilin 2 (PSEN2), on chromosome 1q31–q42 (Ertekin-Taner 2007). APP mutations account for less than 0.1 % of AD patients. Mutations in APP are located near the cleavage sites of the protein resulting in increased production of the Amyloid beta peptide (Aβ). The average age of disease onset for this mutation is between 40s and 50s. The majority of AD cases have complex etiology due to both environmental and genetic factors, which alone do not seem sufficient for causing the disease. Several works are demonstrating direct epigenetic modulation of the disease.
The methylation status of 12 specific genes that have been implicated in AD pathology has been reported to exhibit significant “epigenetic drift.” It was observed in some of the CpG sites within the DNA-methyltransferase 1 (DNMT1) promoter (Mastroeni et al. 2011). Several studies are demonstrating a correlation between dietary factors, the epigenome , and AD pathology. Nutritional deficit could lead to hyperhomocysteinemia due to alteration in the homocysteine/S-adenosylmethionine (Hcy/SAM) (Obeid and Herrmann 2006). Several reports have demonstrated alterations in histone proteins in AD. Phosphorylation of histone H3, a key step in the activation of the mitotic machinery, is increased to a hyperphosphorylated state in AD hippocampal neurons. A nonnuclear form of histone H1 appears to be upregulated in astrocytes and neurons in brain regions that are rich in AD pathology. H1 preferentially binds Aβ-42, as well as Aβ-like structures of numerous proteins (Duce et al. 2006). In addition, the H1 molecule has been shown to be a major target for poly-ADP ribosylation in areas of AD brain.
Aging is universally considered to be one of the most striking risk factors for AD . Why aging should be a risk factor for AD (and other age-related disorders), however, is not well understood, particularly at a mechanistic level. Progressive age-related decline in total methylcytosine has been reported in various organisms. It has been speculated that progressive, age-related, genome-wide hypomethylation may be due to parallel DNMT1 deficits. Age-dependent increase in S-adenosyl-homocysteine (SAH) relative to SAM might also play a role (Mastroeni et al. 2011). Age-dependent hypomethylation of a number of specific genes related to AD has been reported. For example, methylation of cytosines in the APP promoter, particularly GC-rich elements from approximately −270 to −182 bp, is significantly lower in autopsy cases of 70 years old individuals compared to cases with much younger individuals (Mastroeni et al. 2011). These age-related modifications on DNA methylation alter APP expression and consequently can affect the progressive Aβ deposition with aging in the brain (Tohgi et al. 1999).
Parkinson’s disease (PD) is another neurodegenerative disorder associated with aging, affecting the central nervous system and by so effecting the motor functions of the individual producing the so-called Parkinsonian gait characterized by small shuffling steps, dyskinesia presented in the form of hyperkinesia or hypokinesia, and in extreme cases akinesia (Morris et al. 1998). The pathophysiology of the disease is presented as progressive loss of dopaminergic neurons in the substantia nigra (SN) pars compacta and the frequent histological findings of Lewy bodies composed from the aggregation of alpha-synuclein proteins (Weintraub et al. 2008).
The majority of PD onsets are sporadic which accounts for about 90–95 % of the cases, but the causative factors of the disease are far from concluded. There are many models that link genetics, environmental pollution, as well possible contributions of epigenetics to PD. In 5–10 % of the cases of the disease are genetically provoked meaning that the onset of the disease has a genetic factor. Several genes and chromosomal loci, linked to the development of familial forms of Parkinson disease, named PARK1-16, are associated with autosomal dominant, recessive, and X-linked forms of the disease (Hardy et al. 2009). SNCA gene encodes for alpha-synuclein, a protein abundant in brain tissue and when mutated or elevated has the propensity to form the building blocks for the formation of Lewy bodies in PD. Three point mutations and multiplications in the SNCA gene are known to be linked to autosomal dominant PD (Klein and Schlossmacher 2007).
Another gene that is considered to be a factor for both sporadic and familial forms of the disease is LRRK2. The LRRK2 gene encodes for the large-280 kDa leucine-rich repeat kinase multifunctional protein also called dardarin. The most observed mutations in both sporadic and dominant variants of PD are the increase of catalytic activity of dardarin which causes neuronal toxicity and dopaminergic neuron degeneration (Abdullah et al. 2014). Some authors postulate that change in gene transcription exerts a modulating effect on the central nervous system (CNS) and pathophysiology of neurodegenerative diseases. Epigenetic linkage in neurodegenerative diseases is yet inconclusive and more data are needed; however, there is evidence showing the role of DNA methylation in PD and the role of chromatin remodeling in the persisting effects of dopamine on brain function. Strong evidence indicates that DNA methylation in PD is based on the deregulation of homocysteine (Hcy). Generally, the levels of homocysteine (Hcy) are variable among individuals.
This is the result of genetic or environmental factors that influence the levels of dietary folate thus having a major impact on homocysteine (Hcy) levels (Giles et al. 1995). In the metabolism of methylation reactions, folate is converted into l-methylfolate (l-MTHF) by the enzyme dihydrofolate (DHF) reductase in order to be absorbed by the intestine. The next step is the association of l-MTHF with Hcy and methionine synthetase producing methionine. Methionine is converted to S-adenosylmethionine (SAM) and through the joint action of methyltransferases the product S-adenosylhomocysteine (SAH) is formed from which subsequently Hcy is generated. The generated Hcy reenters the methylation cycle and serves for generating methyl groups required for methylation reactions on DNA CpG islands or onto the lysine and arginine residues of histone proteins. Increased concentrations of plasma total Hcy have been reported in patients with PD; this effects the SAM/SAH ratio by increasing SAH and decreasing SAM, leading to an overall decrease in methylation potential (Blandini et al. 2001). Better cognitive functions were associated with increased SAM/SAH ratio in patients with PD. This suggests a possible role of methylation in neurodegenerative disorders like PD (Obeid et al. 2009). There are several studies that submit evidence proving the central role of histone modification in neurotoxicity and thus the role of epigenetics in it.
2.7 Chromatin Regulation of Viral Infection
2.7.1 Differences in Susceptibility Between Species
Whether a pathogen can cause disease in a host is dependent not only on the virulence of the pathogen but also on the genetic background, the health status, and the age of the host. The differences in susceptibility may be related to a number of factors among which the age can have an overall effect on disease resistance, with the very young and the very old being more susceptible to infection by a wide variety of pathogens. Stress in the form of extreme exertion, shock, a change in environment , climate change, nervousness, or muscle fatigue can have a negative impact on health. Each of these conditions is thought to increase the release of cortisol from the adrenal cortex, causing a suppression of the inflammatory response, thereby facilitating infection. Many viruses introduce DNA into the host-cell nucleus, where they must either embrace or confront chromatin factors as a support or obstacle to completion of its life cycle. Compared to the eukaryotic cell, viruses have compact and rapidly evolving genomes.
Chromatin dynamics and epigenetic modifications play major roles in viral and host chromosome biology (Tempera and Lieberman 2014). In some cases, viruses may use novel or viral-specific epigenetic modifying activities, which may reflect variant pathways that distinguish their behavior from the bulk of the cellular chromosome. The role of chromatin in virus biology depends largely on the lifestyle of the virus, but for all viruses that transverse the nucleus, interactions with chromatin are unavoidable. A number of recent discoveries in virology underscore the importance of chromatin dynamics in the regulation of essential viral processes, including entry, gene expression, and replication (Lieberman 2006).
Cellular chromatin forms a dynamic structure that maintains the stability and accessibility of the host DNA genome. Viruses that enter and persist in the nucleus must, therefore, contend with the forces that drive chromatin formation and regulate chromatin structure (Lieberman 2006; Robertson 2005). The complex and dynamic properties of nuclear chromatin present a formidable challenge to viral gene expression and genome propagation. Recently, a wealth of information has been uncovered regarding the structure, function, and regulation of chromatin during complex cellular processes such as differentiation , recombination, aging, and carcinogenesis. Recent studies have also revealed that chromatin has a major role in the life cycle of many viruses, and that viruses have coevolved with numerous strategies for modulating chromatin-related processes (Lieberman 2006). The genomes of many DNA viruses persist for considerable lengths of time in the host-cell nucleus. The small DNA tumor viruses such as simian virus 40 (SV40) and polyoma virus are assembled into nucleosomal minichromosomes during the DNA replication process. Because these viruses use cellular replication enzymes, it is thought that the viral minichromosomes are assembled through a mechanism that is indistinguishable from nucleosome assembly on replicating cellular chromosomes (Lieberman 2006).
Most viruses package their nucleic acid genomes at very high molecular density with specialized viral packaging proteins to form a capsid particle. Adenovirus genomes are packaged as linear double stranded DNA with viral core proteins that form a chromatin -like structure. The viral DNA is covalently linked with the viral encoded terminal binding protein (TP) and core proteins VII, V, and X (Haruki et al. 2006). Core protein VII has sequence similarity to histone H3 and the basic sperm-specific protein. Transcription of the newly infecting genome utilizes a template that decondenses during entry into the nucleus. The cellular activator, referred to as the Template Activating Factor (TAF), is required for the replication competence of the viral DNA (Haruki et al. 2006).
Herpes Simplex Virus (HSV), like adenovirus, enters cells as a linear double stranded DNA molecule. Several virion-associated proteins are essential for productive infection. The HSV VP22 protein binds to TAF1B, similar to that of Ad VII protein (van Leeuwen et al. 2003). VP16 is another HSV-encoded virion protein that plays a very active role in transcription activation of the viral immediate early genes. VP16 recruits histone modifying and nucleosome remodeling enzymes which indicate that nucleosomes are assembling onto the viral genome during viral entry into the nucleus. The precise details of nucleosome assembly during the early stages of herpes virus entry have not been completely elucidated (Lieberman 2008).
Retrovirus and lentivirus genomes also enter the nucleus as double stranded DNA, but this process appears to differ significantly from that of the constitutively double stranded nuclear DNA viruses, like adeno- and herpes viruses. One major difference is that the double stranded DNA genome is synthesized by virion-associated reverse transcriptase in the cytoplasm. The newly synthesized cytoplasmic DNA forms a pre-integration complex (PIC) that consists of viral and cellular proteins (Suzuki and Craigie 2007). Entry into the nucleus is cell cycle dependent and utilizes the cellular nuclear import machinery, often through a nuclear localization signal on one or more of the PIC components. Once in the nucleus, PIC components like Lens Epithelium-Derived Growth Factor, (LEDGF) have been implicating in anchoring to chromatin . HIV, for example, was found to integrate at chromatin sites enriched in euchromatic histone modifications, including H3 K4 methylation and histone H3 and H4 acetylation (Wang et al. 2007). LEDGF appears to be a decisive and essential factor in mediating the chromatin attachment of the PIC and viral integration (Llano et al. 2006).
The role of chromatin in the organization of latent viruses may help to provide insight into genome stability in general. Viruses, although smaller than cellular chromosomes, confront many of the same challenges as the cellular chromosome. How viruses manage these tasks in the absence of canonical centromeres and telomeres, and sometimes lacking dedicated origins of DNA replication, will be important to define in the near future (Lieberman 2008).
Viruses may trigger in the infected target cells a pathway of programmed cell death (apoptosis), which is characterized by distinct morphological changes such as the formation of condensed fragments of nuclear chromatin . Apoptosis is an energy-dependent process that can be triggered from the activation of calcium-dependent endonucleases whose function is to cleave genomic DNA. The phenomenon of apoptosis seems to play an important role in the control of virus infection since several viruses have developed ways to counteract the apoptosis. Herpes virus, Epstein–Barr virus, adenoviruses, and African swine fever virus have all been reported to code a protein that functions as an inhibitor of apoptosis (Ackermann et al. 2000).
2.7.2 Age-Dependent Organism Susceptibility to Viral Infections
Deterioration of the immune system function is common in advanced aging. Older people experience enhanced susceptibility to infections. This susceptibility is due to many factors but also due to decline in the immune response. The immune system is the adaptive defense system in vertebrates, which evolved to protect them from invading pathogenic microorganisms, viruses, and cancer . It has the ability to generate a variety of immuno-competent cells and molecules that specifically recognize and eliminate pathogens.
Older organisms may succumb to viral infection due to exaggerated immune response as it is proven that in old mouse models there is an elevated immune response as aged mice produce higher serum levels of inflammatory mediator IL-17 than their younger counterparts upon herpes virus infection. Major contributor for the elevated IL-17 are the NKT cells in the aged mice. Aging induces increase in RORyT transcription factor within NKT cell promoter of IL-17. During herpes virus infection, these aged cells produce exaggerated levels of IL-17 leading to worse outcomes in aged host. This challenges the more accepted notion that reduced immunity with aging is the dominant reason for the aged hosts to be more susceptible to viral infections. In unison is the notion that aged, but not young, mice succumb to systemic herpes viral infection due to exaggerated inflammation (Goldstein 2010).
Advanced age also increases the susceptibility of the organism to influenza infection. The mechanism underlying the impaired immune response to infection is not well understood. There are data showing that the advanced age affects dendritic cells’ (DC) functions. It has also been shown that the monocyte-derived DC cells from the aged organism are impaired in their capacity to secrete IFN-1 and INF-3, both playing a role in organismal defense against viral respiratory infections. Some authors advocate that the reduction of IFN-1 and IFN-3 is a result of age-associated modifications in the chromatin structure, namely the increase in H3K4me3 and H3K9me3 at the promoters of IFN-1 and IFN-3 (Prakash et al. 2013). This was accompanied by decreased association of these promoters with activator histone marks like H3K4me3 in aged DCs after activation with influenza, thus highlighting the significance of chromatin organization for the age-dependent susceptibility of organisms to viral infections.
2.8 Conclusion
Knowing the intricate mechanisms of aging, age-associated diseases, premature aging syndromes, and the dependence of organismal susceptibility to viruses on aging is a challenge for scientists. It could open new horizons in the field of pharmacogenomics and moreover in the field of yet not-developed pharmacoepigenomics. The last is going to be on the top of the iceberg called “personalized medicine” as it offers brilliant opportunities for scientists to know, control, and heal all age-associated epigenetic changes in the organisms.
References
Abdullah, R., Basak, I., Patil, K. S., Alves, G., Larsen, J. P., & Moller, S. G. (2014). Parkinson’s disease and age: The obvious but largely unexplored link. Experimental Gerontology. doi:10.1016/j.exger.2014.09.014.
Ackermann, H. W., Tremblay, M., & Berthiaume, L. (2000). Viral pathogenesis in diagrams. Boca Raton, FL: CRC Press.
Adams, P. D. (2007). Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene, 397(1–2), 84–93. doi:10.1016/j.gene.2007.04.020.
Ahuja, N., Li, Q., Mohan, A. L., Baylin, S. B., & Issa, J. P. (1998). Aging and DNA methylation in colorectal mucosa and cancer. Cancer Research, 58(23), 5489–5494.
Andersson, K., Mahr, R., Bjorkroth, B., & Daneholt, B. (1982). Rapid reformation of the thick chromosome fiber upon completion of RNA synthesis at the Balbiani ring genes in Chironomus tentans. Chromosoma, 87(1), 33–48.
Apostolou, E., & Hochedlinger, K. (2013). Chromatin dynamics during cellular reprogramming. Nature, 502(7472), 462–471. doi:10.1038/nature12749.
Ashraf, N., Zino, S., Macintyre, A., Kingsmore, D., Payne, A. P., George, W. D., et al. (2006). Altered sirtuin expression is associated with node-positive breast cancer. British Journal of Cancer, 95(8), 1056–1061. doi:10.1038/sj.bjc.6603384.
Bannister, A. J., & Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Research, 21(3), 381–395. doi:10.1038/cr.2011.22.
Barlesi, F., Giaccone, G., Gallegos-Ruiz, M. I., Loundou, A., Span, S. W., Lefesvre, P., et al. (2007). Global histone modifications predict prognosis of resected non small-cell lung cancer. Journal of Clinical Oncology, 25(28), 4358–4364. doi:10.1200/JCO.2007.11.2599.
Behbahani, T. E., Kahl, P., von der Gathen, J., Heukamp, L. C., Baumann, C., Gutgemann, I., et al. (2012). Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urology, 12, 5. doi:10.1186/1471-2490-12-5.
Bender, S., Tang, Y., Lindroth, A. M., Hovestadt, V., Jones, D. T., Kool, M., et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell, 24(5), 660–672. doi:10.1016/j.ccr.2013.10.006.
Benetti, R., Garcia-Cao, M., & Blasco, M. A. (2007a). Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nature Genetics, 39(2), 243–250. doi:10.1038/ng1952.
Benetti, R., Gonzalo, S., Jaco, I., Schotta, G., Klatt, P., Jenuwein, T., et al. (2007b). Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. The Journal of Cell Biology, 178(6), 925–936. doi:10.1083/jcb.200703081.
Bettens, K., Sleegers, K., & Van Broeckhoven, C. (2013). Genetic insights in Alzheimer’s disease. The Lancet Neurology, 12(1), 92–104. doi:10.1016/S1474-4422(12)70259-4.
Bianco-Miotto, T., Chiam, K., Buchanan, G., Jindal, S., Day, T. K., Thomas, M., et al. (2010). Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiology, Biomarkers & Prevention, 19(10), 2611–2622. doi:10.1158/1055-9965.EPI-10-0555.
Blandini, F., Fancellu, R., Martignoni, E., Mangiagalli, A., Pacchetti, C., Samuele, A., et al. (2001). Plasma homocysteine and l-dopa metabolism in patients with Parkinson disease. Clinical Chemistry, 47(6), 1102–1104.
Blasco, M. A. (2007). The epigenetic regulation of mammalian telomeres. Nature Reviews Genetics, 8(4), 299–309. doi:10.1038/nrg2047.
Bowen, N. J., Fujita, N., Kajita, M., & Wade, P. A. (2004). Mi-2/NuRD: Multiple complexes for many purposes. Biochimica et Biophysica Acta, 1677(1-3), 52–57. doi:10.1016/j.bbaexp.2003.10.010.
Burton, A., & Torres-Padilla, M. E. (2014). Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nature Reviews Molecular Cell Biology, 15(11), 723–734. doi:10.1038/nrm3885.
Campion, D., Dumanchin, C., Hannequin, D., Dubois, B., Belliard, S., Puel, M., et al. (1999). Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. American Journal of Human Genetics, 65(3), 664–670. doi:10.1086/302553.
Campisi, J. (2000). Cancer, aging and cellular senescence. In Vivo, 14(1), 183–188.
Cao, K., Blair, C. D., Faddah, D. A., Kieckhaefer, J. E., Olive, M., Erdos, M. R., et al. (2011). Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. The Journal of Clinical Investigation, 121(7), 2833–2844. doi:10.1172/JCI43578.
Casillas, M. A., Jr., Lopatina, N., Andrews, L. G., & Tollefsbol, T. O. (2003). Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Molecular and Cellular Biochemistry, 252(1–2), 33–43.
Catez, F., Ueda, T., & Bustin, M. (2006). Determinants of histone H1 mobility and chromatin binding in living cells. Nature Structural & Molecular Biology, 13(4), 305–310. doi:10.1038/nsmb1077.
Chandra, T., Ewels, P. A., Schoenfelder, S., Furlan-Magaril, M., Wingett, S. W., Kirschner, K., et al. (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Reports, 10(4), 471–484. doi:10.1016/j.celrep.2014.12.055.
Chen, X., Sun, K., Jiao, S., Cai, N., Zhao, X., Zou, H., et al. (2014a). High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Scientific Reports, 4, 7481. doi:10.1038/srep07481.
Chen, Z. H., Zhu, M., Yang, J., Liang, H., He, J., He, S., et al. (2014b). PTEN interacts with histone H1 and controls chromatin condensation. Cell Reports, 8(6), 2003–2014. doi:10.1016/j.celrep.2014.08.008.
Cheung, I., Shulha, H. P., Jiang, Y., Matevossian, A., Wang, J., Weng, Z., et al. (2010). Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America, 107(19), 8824–8829. doi:10.1073/pnas.1001702107.
Cho, K. S., Elizondo, L. I., & Boerkoel, C. F. (2004). Advances in chromatin remodeling and human disease. Current Opinion in Genetics & Development, 14(3), 308–315. doi:10.1016/j.gde.2004.04.015.
Chuang, C. H., Carpenter, A. E., Fuchsova, B., Johnson, T., de Lanerolle, P., & Belmont, A. S. (2006). Long-range directional movement of an interphase chromosome site. Current Biology, 16(8), 825–831. doi:10.1016/j.cub.2006.03.059.
Clapier, C. R., & Cairns, B. R. (2009). The biology of chromatin remodeling complexes. Annual Review of Biochemistry, 78, 273–304. doi:10.1146/annurev.biochem.77.062706.153223.
Clausell, J., Happel, N., Hale, T. K., Doenecke, D., & Beato, M. (2009). Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-dependent remodeling by SWI/SNF or NURF. PLoS One, 4(10), e0007243. doi:10.1371/journal.pone.0007243.
Columbaro, M., Capanni, C., Mattioli, E., Novelli, G., Parnaik, V. K., Squarzoni, S., et al. (2005). Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cellular and Molecular Life Sciences, 62(22), 2669–2678. doi:10.1007/s00018-005-5318-6.
Cook, P. R., & Brazell, I. A. (1975). Supercoils in human DNA. Journal of Cell Science, 19(2), 261–279.
Corona, D. F., Siriaco, G., Armstrong, J. A., Snarskaya, N., McClymont, S. A., Scott, M. P., et al. (2007). ISWI regulates higher-order chromatin structure and histone H1 assembly in vivo. PLoS Biology, 5(9), e232. doi:10.1371/journal.pbio.0050232.
Corpet, A., & Stucki, M. (2014). Chromatin maintenance and dynamics in senescence: A spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma, 123(5), 423–436. doi:10.1007/s00412-014-0469-6.
Cremer, T., & Cremer, C. (2006). Rise, fall and resurrection of chromosome territories: a historical perspective. Part II. Fall and resurrection of chromosome territories during the 1950s to 1980s. Part III. Chromosome territories and the functional nuclear architecture: Experiments and models from the 1990s to the present. European Journal of Histochemistry, 50(4), 223–272.
Cuthbert, G. L., Daujat, S., Snowden, A. W., Erdjument-Bromage, H., Hagiwara, T., Yamada, M., et al. (2004). Histone deimination antagonizes arginine methylation. Cell, 118(5), 545–553. doi:10.1016/j.cell.2004.08.020.
Dalvai, M., & Bystricky, K. (2010). The role of histone modifications and variants in regulating gene expression in breast cancer. Journal of Mammary Gland Biology and Neoplasia, 15(1), 19–33. doi:10.1007/s10911-010-9167-z.
Dang, W., Steffen, K. K., Perry, R., Dorsey, J. A., Johnson, F. B., Shilatifard, A., et al. (2009). Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature, 459(7248), 802–807. doi:10.1038/nature08085.
De Sandre-Giovannoli, A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I., et al. (2003). Lamin a truncation in Hutchinson-Gilford progeria. Science, 300(5628), 2055. doi:10.1126/science.1084125.
Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shumaker, D. K., Solimando, L., et al. (2008). Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes & Development, 22(7), 832–853. doi:10.1101/gad.1652708.
Decker, M. L., Chavez, E., Vulto, I., & Lansdorp, P. M. (2009). Telomere length in Hutchinson-Gilford progeria syndrome. Mechanisms of Ageing and Development, 130(6), 377–383. doi:10.1016/j.mad.2009.03.001.
Dekker, J. (2008). Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. The Journal of Biological Chemistry, 283(50), 34532–34540. doi:10.1074/jbc.M806479200.
Dekker, J., Rippe, K., Dekker, M., & Kleckner, N. (2002). Capturing chromosome conformation. Science, 295(5558), 1306–1311. doi:10.1126/science.1067799.
Deligezer, U., Akisik, E. Z., Akisik, E. E., Kovancilar, M., Bugra, D., Erten, N., Holdenrieder, S., & Dalay, N. (2011). H3K9me3/H4K20me3 ratio in circulating nucleosomes as potential biomarker for colorectal cancer. In P. B. Gahan (Ed.), Circulating nucleic acids in plasma and serum (pp. 97–103). Dordrecht: Springer.
Dhanasekaran, K., Arif, M., & Kundu, T. K. (2012). Cancer: An epigenetic landscape. In T. K. Kundu (Ed.), Epigenetics: Development and disease (Subcellular Biochemistry, Vol. 61). Netherlands: Springer.
Dhanasekaran, K., Kumari, S., & Kanduri, C. (2013). Noncoding RNAs in chromatin organization and transcription regulation: An epigenetic view. Sub-Cellular Biochemistry, 61, 343–372. doi:10.1007/978-94-007-4525-4_15.
Dorigo, B., Schalch, T., Bystricky, K., & Richmond, T. J. (2003). Chromatin fiber folding: Requirement for the histone H4 N-terminal tail. Journal of Molecular Biology, 327(1), 85–96.
Downs, J. A., & Cote, J. (2005). Dynamics of chromatin during the repair of DNA double-strand breaks. Cell Cycle, 4(10), 1373–1376. doi:10.4161/cc.4.10.2108.
Downs, J. A., Kosmidou, E., Morgan, A., & Jackson, S. P. (2003). Suppression of homologous recombination by the Saccharomyces cerevisiae linker histone. Molecular Cell, 11(6), 1685–1692.
Duce, J. A., Smith, D. P., Blake, R. E., Crouch, P. J., Li, Q. X., Masters, C. L., et al. (2006). Linker histone H1 binds to disease associated amyloid-like fibrils. Journal of Molecular Biology, 361(3), 493–505. doi:10.1016/j.jmb.2006.06.038.
Earnshaw, W. C., & Laemmli, U. K. (1983). Architecture of metaphase chromosomes and chromosome scaffolds. The Journal of Cell Biology, 96(1), 84–93.
Ehrlich, M. (2009). DNA hypomethylation in cancer cells. Epigenomics, 1(2), 239–259. doi:10.2217/epi.09.33.
Ellinger, J., Kahl, P., Mertens, C., Rogenhofer, S., Hauser, S., Hartmann, W., et al. (2010a). Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. International Journal of Cancer, 127(10), 2360–2366. doi:10.1002/ijc.25250.
Ellinger, J., Kahl, P., von der Gathen, J., Heukamp, L. C., Gutgemann, I., Walter, B., et al. (2012). Global histone H3K27 methylation levels are different in localized and metastatic prostate cancer. Cancer Investigation, 30(2), 92–97. doi:10.3109/07357907.2011.636117.
Ellinger, J., Kahl, P., von der Gathen, J., Rogenhofer, S., Heukamp, L. C., Gutgemann, I., et al. (2010b). Global levels of histone modifications predict prostate cancer recurrence. The Prostate, 70(1), 61–69. doi:10.1002/pros.21038.
Elsheikh, S. E., Green, A. R., Rakha, E. A., Powe, D. G., Ahmed, R. A., Collins, H. M., et al. (2009). Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Research, 69(9), 3802–3809. doi:10.1158/0008-5472.CAN-08-3907.
Eriksson, M., Brown, W. T., Gordon, L. B., Glynn, M. W., Singer, J., Scott, L., et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature, 423(6937), 293–298. doi:10.1038/nature01629.
Ertekin-Taner, N. (2007). Genetics of Alzheimer’s disease: A centennial review. Neurologic Clinics, 25(3), 611–667. doi:10.1016/j.ncl.2007.03.009. v.
Fan, Y., Nikitina, T., Zhao, J., Fleury, T. J., Bhattacharyya, R., Bouhassira, E. E., et al. (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell, 123(7), 1199–1212. doi:10.1016/j.cell.2005.10.028.
Feser, J., & Tyler, J. (2011). Chromatin structure as a mediator of aging. FEBS Letters, 585(13), 2041–2048. doi:10.1016/j.febslet.2010.11.016.
Filion, G. J., van Bemmel, J. G., Braunschweig, U., Talhout, W., Kind, J., Ward, L. D., et al. (2010). Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell, 143(2), 212–224. doi:10.1016/j.cell.2010.09.009.
Finch, J. T., & Klug, A. (1976). Solenoidal model for superstructure in chromatin. Proceedings of the National Academy of Sciences of the United States of America, 73(6), 1897–1901.
Fraga, M. F., Agrelo, R., & Esteller, M. (2007). Cross-talk between aging and cancer: The epigenetic language. Annals of the New York Academy of Sciences, 1100, 60–74. doi:10.1196/annals.1395.005.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005a). Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America, 102(30), 10604–10609. doi:10.1073/pnas.0500398102.
Fraga, M. F., Ballestar, E., Villar-Garea, A., Boix-Chornet, M., Espada, J., Schotta, G., et al. (2005b). Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics, 37(4), 391–400. doi:10.1038/ng1531.
Fraser, P. (2006). Transcriptional control thrown for a loop. Current Opinion in Genetics & Development, 16(5), 490–495. doi:10.1016/j.gde.2006.08.002.
Fuke, C., Shimabukuro, M., Petronis, A., Sugimoto, J., Oda, T., Miura, K., et al. (2004). Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: An HPLC-based study. Annals of Human Genetics, 68(Pt 3), 196–204. doi:10.1046/j.1529-8817.2004.00081.x.
Fullgrabe, J., Kavanagh, E., & Joseph, B. (2011). Histone onco-modifications. Oncogene, 30(31), 3391–3403. doi:10.1038/onc.2011.121.
Funayama, R., Saito, M., Tanobe, H., & Ishikawa, F. (2006). Loss of linker histone H1 in cellular senescence. The Journal of Cell Biology, 175(6), 869–880. doi:10.1083/jcb.200604005.
Fussner, E., Strauss, M., Djuric, U., Li, R., Ahmed, K., Hart, M., et al. (2012). Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Reports, 13(11), 992–996. doi:10.1038/embor.2012.139.
Gabrovsky, N., Georgieva, M., Laleva, M., Uzunov, K., & Miloshev, G. (2013). Histone H1.0–a potential molecular marker with prognostic value for patients with malignant gliomas. Acta Neurochirurgica, 155(8), 1437–1442. doi:10.1007/s00701-013-1802-1.
Garinis, G. A., van der Horst, G. T., Vijg, J., & Hoeijmakers, J. H. (2008). DNA damage and ageing: New-age ideas for an age-old problem. Nature Cell Biology, 10(11), 1241–1247. doi:10.1038/ncb1108-1241.
Gasser, S. M. (2002). Visualizing chromatin dynamics in interphase nuclei. Science, 296(5572), 1412–1416. doi:10.1126/science.1067703.
Gelato, K. A., & Fischle, W. (2008). Role of histone modifications in defining chromatin structure and function. Biological Chemistry, 389(4), 353–363. doi:10.1515/BC.2008.048.
Georgieva, M., Harata, M., & Miloshev, G. (2008). The nuclear actin-related protein Act3p/Arp4 influences yeast cell shape and bulk chromatin organization. Journal of Cellular Biochemistry, 104(1), 59–67. doi:10.1002/jcb.21600.
Georgieva, M., Roguev, A., Balashev, K., Zlatanova, J., & Miloshev, G. (2012). Hho1p, the linker histone of Saccharomyces cerevisiae, is important for the proper chromatin organization in vivo. Biochimica et Biophysica Acta, 1819(5), 366–374. doi:10.1016/j.bbagrm.2011.12.003.
Georgieva, M., Staneva, D., Uzunova, K., Efremov, T., Balashev, K., Harata, M., et al. (2015). The linker histone in Saccharomyces cerevisiae interacts with actin-related protein 4 and both regulate chromatin structure and cellular morphology. The International Journal of Biochemistry & Cell Biology, 59, 182–192. doi:10.1016/j.biocel.2014.12.006.
Ghirlando R, Felsenfeld G (2013) Chromatin structure outside and inside the nucleus. Biopolymers 99: 225–232.
Gilbert, N., Boyle, S., Fiegler, H., Woodfine, K., Carter, N. P., & Bickmore, W. A. (2004). Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell, 118(5), 555–566. doi:10.1016/j.cell.2004.08.011.
Giles, W. H., Kittner, S. J., Anda, R. F., Croft, J. B., & Casper, M. L. (1995). Serum folate and risk for ischemic stroke. First National Health and Nutrition Examination Survey epidemiologic follow-up study. Stroke, 26(7), 1166–1170.
Goldman, R. D., Gruenbaum, Y., Moir, R. D., Shumaker, D. K., & Spann, T. P. (2002). Nuclear lamins: Building blocks of nuclear architecture. Genes & Development, 16(5), 533–547. doi:10.1101/gad.960502.
Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M., Goldman, A. E., Gordon, L. B., et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proceedings of the National Academy of Sciences of the United States of America, 101(24), 8963–8968. doi:10.1073/pnas.0402943101.
Goldstein, D. R. (2010). Aging, imbalanced inflammation and viral infection. Virulence, 1(4), 295–298. doi:10.4161/viru.1.4.12009.
Gordon, L. B., Rothman, F. G., Lopez-Otin, C., & Misteli, T. (2014). Progeria: A paradigm for translational medicine. Cell, 156(3), 400–407. doi:10.1016/j.cell.2013.12.028.
Gotta, M., Strahl-Bolsinger, S., Renauld, H., Laroche, T., Kennedy, B. K., Grunstein, M., et al. (1997). Localization of Sir2p: The nucleolus as a compartment for silent information regulators. The EMBO Journal, 16(11), 3243–3255. doi:10.1093/emboj/16.11.3243.
Gravina, S., & Vijg, J. (2010). Epigenetic factors in aging and longevity. Pflügers Archiv - European Journal of Physiology, 459(2), 247–258. doi:10.1007/s00424-009-0730-7.
Grigoryev, S. A., Arya, G., Correll, S., Woodcock, C. L., & Schlick, T. (2009). Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proceedings of the National Academy of Sciences of the United States of America, 106(32), 13317–13322. doi:10.1073/pnas.0903280106.
Guarente, L. (2013). Introduction: Sirtuins in aging and diseases. Methods in Molecular Biology, 1077, 3–10. doi:10.1007/978-1-62703-637-5_1.
Guerrero, N., Mendes de Leon, C. F., Evans, D. A., & Jacobs, E. A. (2015). Determinants of trust in health care in an older population. Journal of the American Geriatrics Society. doi:10.1111/jgs.13316.
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell, 49(2), 359–367. doi:10.1016/j.molcel.2012.10.016.
Happel, N., & Doenecke, D. (2009). Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene, 431(1-2), 1–12. doi:10.1016/j.gene.2008.11.003.
Hardy, J., Lewis, P., Revesz, T., Lees, A., & Paisan-Ruiz, C. (2009). The genetics of Parkinson’s syndromes: A critical review. Current Opinion in Genetics & Development, 19(3), 254–265. doi:10.1016/j.gde.2009.03.008.
Haruki, H., Okuwaki, M., Miyagishi, M., Taira, K., & Nagata, K. (2006). Involvement of template-activating factor I/SET in transcription of adenovirus early genes as a positive-acting factor. Journal of Virology, 80(2), 794–801. doi:10.1128/JVI.80.2.794-801.2006.
Hayes, J. J., Tullius, T. D., & Wolffe, A. P. (1990). The structure of DNA in a nucleosome. Proceedings of the National Academy of Sciences of the United States of America, 87(19), 7405–7409.
He, C., Xu, J., Zhang, J., Xie, D., Ye, H., Xiao, Z., et al. (2012). High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Human Pathology, 43(9), 1425–1435. doi:10.1016/j.humpath.2011.11.003.
Hegele, R. (2005). LMNA mutation position predicts organ system involvement in laminopathies. Clinical Genetics, 68(1), 31–34. doi:10.1111/j.1399-0004.2005.00447.x.
Helbling Chadwick, L., Chadwick, B. P., Jaye, D. L., & Wade, P. A. (2009). The Mi-2/NuRD complex associates with pericentromeric heterochromatin during S phase in rapidly proliferating lymphoid cells. Chromosoma, 118(4), 445–457. doi:10.1007/s00412-009-0207-7.
Henderson, K. A., & Hughes, A. L. (2014). Mother-daughter asymmetry of pH underlies aging and rejuvenation in yeast. Elife, 3, e03504. doi:10.7554/eLife.03504.
Hennekam, R. C. (2006). Hutchinson-Gilford progeria syndrome: Review of the phenotype. American Journal of Medical Genetics Part A, 140(23), 2603–2624. doi:10.1002/ajmg.a.31346.
Hermann, A., Gowher, H., & Jeltsch, A. (2004). Biochemistry and biology of mammalian DNA methyltransferases. Cellular and Molecular Life Sciences, 61(19–20), 2571–2587. doi:10.1007/s00018-004-4201-1.
Herranz, D., & Serrano, M. (2010). SIRT1: Recent lessons from mouse models. Nature Reviews Cancer, 10(12), 819–823. doi:10.1038/nrc2962.
Herskind, A. M., McGue, M., Holm, N. V., Sorensen, T. I., Harvald, B., & Vaupel, J. W. (1996). The heritability of human longevity: A population-based study of 2872 Danish twin pairs born 1870-1900. Human Genetics, 97(3), 319–323.
Herskovits, A. Z., & Guarente, L. (2014). SIRT1 in neurodevelopment and brain senescence. Neuron, 81(3), 471–483. doi:10.1016/j.neuron.2014.01.028.
Heyn, H., Moran, S., & Esteller, M. (2013). Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics, 8(1), 28–33. doi:10.4161/epi.23366.
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14(10), R115. doi:10.1186/gb-2013-14-10-r115.
Horvath, S., Garagnani, P., Bacalini, M. G., Pirazzini, C., Salvioli, S., Gentilini, D., et al. (2015). Accelerated epigenetic aging in Down syndrome. Aging Cell. doi:10.1111/acel.12325.
Huang, S., Risques, R. A., Martin, G. M., Rabinovitch, P. S., & Oshima, J. (2008). Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Experimental Cell Research, 314(1), 82–91. doi:10.1016/j.yexcr.2007.08.004.
Huang, J. C., Yan, L. Y., Lei, Z. L., Miao, Y. L., Shi, L. H., Yang, J. W., et al. (2007). Changes in histone acetylation during postovulatory aging of mouse oocyte. Biology of Reproduction, 77(4), 666–670. doi:10.1095/biolreprod.107.062703.
Huynh, V. A., Robinson, P. J., & Rhodes, D. (2005). A method for the in vitro reconstitution of a defined “30 nm” chromatin fibre containing stoichiometric amounts of the linker histone. Journal of Molecular Biology, 345(5), 957–968. doi:10.1016/j.jmb.2004.10.075.
Inoue, T., Hiratsuka, M., Osaki, M., & Oshimura, M. (2007). The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle, 6(9), 1011–1018.
Ishimi, Y., Kojima, M., Takeuchi, F., Miyamoto, T., Yamada, M., & Hanaoka, F. (1987). Changes in chromatin structure during aging of human skin fibroblasts. Experimental Cell Research, 169(2), 458–467.
Issa, J. P., Ahuja, N., Toyota, M., Bronner, M. P., & Brentnall, T. A. (2001). Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Research, 61(9), 3573–3577.
Issa, J. P., Ottaviano, Y. L., Celano, P., Hamilton, S. R., Davidson, N. E., & Baylin, S. B. (1994). Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nature Genetics, 7(4), 536–540. doi:10.1038/ng0894-536.
Ito, T. (2007). Role of histone modification in chromatin dynamics. Journal of Biochemistry, 141(5), 609–614. doi:10.1093/jb/mvm091.
Iyer, L. M., Abhiman, S., & Aravind, L. (2011). Natural history of eukaryotic DNA methylation systems. Progress in Molecular Biology and Translational Science, 101, 25–104. doi:10.1016/b978-0-12-387685-0.00002-0.
Kadauke, S., & Blobel, G. A. (2009). Chromatin loops in gene regulation. Biochimica et Biophysica Acta, 1789(1), 17–25. doi:10.1016/j.bbagrm.2008.07.002.
Kaeberlein, M., Burtner, C. R., & Kennedy, B. K. (2007). Recent developments in yeast aging. PLoS Genetics, 3(5), e84. doi:10.1371/journal.pgen.0030084.
Karymov, M. A., Tomschik, M., Leuba, S. H., Caiafa, P., & Zlatanova, J. (2001). DNA methylation-dependent chromatin fiber compaction in vivo and in vitro: Requirement for linker histone. FASEB Journal, 15(14), 2631–2641. doi:10.1096/fj.01-0345com.
Kawakami, K., Nakamura, A., Ishigami, A., Goto, S., & Takahashi, R. (2009). Age-related difference of site-specific histone modifications in rat liver. Biogerontology, 10(4), 415–421. doi:10.1007/s10522-008-9176-0.
Kennedy, B. K., Gotta, M., Sinclair, D. A., Mills, K., McNabb, D. S., Murthy, M., et al. (1997). Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell, 89(3), 381–391.
Khare, S. P., Sharma, A., Deodhar, K. K., & Gupta, S. (2011). Overexpression of histone variant H2A.1 and cellular transformation are related in N-nitrosodiethylamine-induced sequential hepatocarcinogenesis. Experimental Biology and Medicine, 236(1), 30–35. doi:10.1258/ebm.2010.010140.
Kim, J. Y., Tavare, S., & Shibata, D. (2005). Counting human somatic cell replications: Methylation mirrors endometrial stem cell divisions. Proceedings of the National Academy of Sciences of the United States of America, 102(49), 17739–17744. doi:10.1073/pnas.0503976102.
Klein, C., & Schlossmacher, M. G. (2007). Parkinson disease, 10 years after its genetic revolution: Multiple clues to a complex disorder. Neurology, 69(22), 2093–2104. doi:10.1212/01.wnl.0000271880.27321.a7.
Kohlmaier, A., Savarese, F., Lachner, M., Martens, J., Jenuwein, T., & Wutz, A. (2004). A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biology, 2(7), E171. doi:10.1371/journal.pbio.0020171.
Kudlow, B. A., Kennedy, B. K., & Monnat, R. J., Jr. (2007). Werner and Hutchinson-Gilford progeria syndromes: Mechanistic basis of human progeroid diseases. Nature Reviews Molecular Cell Biology, 8(5), 394–404. doi:10.1038/nrm2161.
Kuzmichev, A., Nishioka, K., Erdjument-Bromage, H., Tempst, P., & Reinberg, D. (2002). Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes & Development, 16(22), 2893–2905. doi:10.1101/gad.1035902.
Kuzumaki, N., Ikegami, D., Tamura, R., Sasaki, T., Niikura, K., Narita, M., et al. (2010). Hippocampal epigenetic modification at the doublecortin gene is involved in the impairment of neurogenesis with aging. Synapse, 64(8), 611–616. doi:10.1002/syn.20768.
Lambert, J. C., & Amouyel, P. (2007). Genetic heterogeneity of Alzheimer’s disease: Complexity and advances. Psychoneuroendocrinology, 32(Suppl 1), S62–70. doi:10.1016/j.psyneuen.2007.05.015.
Langmore, J. P., & Schutt, C. (1980). The higher order structure of chicken erythrocyte chromosomes in vivo. Nature, 288(5791), 620–622.
Lazarus, A., Banerjee, K. K., & Kolthur-Seetharam, U. (2013). Small changes, big effects: Chromatin goes aging. Sub-Cellular Biochemistry, 61, 151–176. doi:10.1007/978-94-007-4525-4_8.
Leszinski, G., Gezer, U., Siegele, B., Stoetzer, O., & Holdenrieder, S. (2012). Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Research, 32(5), 2199–2205.
Li, L. C. (2014). Chromatin remodeling by the small RNA machinery in mammalian cells. Epigenetics, 9(1), 45–52. doi:10.4161/epi.26830.
Lieberman, P. M. (2006). Chromatin regulation of virus infection. Trends in Microbiology, 14(3), 132–140. doi:10.1016/j.tim.2006.01.001.
Lieberman, P. M. (2008). Chromatin organization and virus gene expression. Journal of Cellular Physiology, 216(2), 295–302. doi:10.1002/jcp.21421.
Lindstrom, D. L., & Gottschling, D. E. (2009). The mother enrichment program: A genetic system for facile replicative life span analysis in Saccharomyces cerevisiae. Genetics, 183(2), 413–422. doi:10.1534/genetics.109.106229.
Lindstrom, D. L., Leverich, C. K., Henderson, K. A., & Gottschling, D. E. (2011). Replicative age induces mitotic recombination in the ribosomal RNA gene cluster of Saccharomyces cerevisiae. PLoS Genetics, 7(3), e1002015. doi:10.1371/journal.pgen.1002015.
Littau, V. C., Burdick, C. J., Allfrey, V. G., & Mirsky, S. A. (1965). The role of histones in the maintenance of chromatin structure. Proceedings of the National Academy of Sciences of the United States of America, 54(4), 1204–1212.
Liu, B., Wang, J., Chan, K. M., Tjia, W. M., Deng, W., Guan, X., et al. (2005). Genomic instability in laminopathy-based premature aging. Nature Medicine, 11(7), 780–785. doi:10.1038/nm1266.
Liu, Y., Xie, Q. R., Wang, B., Shao, J., Zhang, T., Liu, T., et al. (2013). Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein & Cell. doi:10.1007/s13238-013-3054-5.
Llano, M., Saenz, D. T., Meehan, A., Wongthida, P., Peretz, M., Walker, W. H., et al. (2006). An essential role for LEDGF/p75 in HIV integration. Science, 314(5798), 461–464. doi:10.1126/science.1132319.
Longo, V. D., & Fabrizio, P. (2012). Chronological aging in Saccharomyces cerevisiae. Sub-Cellular Biochemistry, 57, 101–121. doi:10.1007/978-94-007-2561-4_5.
Longo, V. D., & Kennedy, B. K. (2006). Sirtuins in aging and age-related disease. Cell, 126(2), 257–268. doi:10.1016/j.cell.2006.07.002.
Lopatina, N., Haskell, J. F., Andrews, L. G., Poole, J. C., Saldanha, S., & Tollefsbol, T. (2002). Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. Journal of Cellular Biochemistry, 84(2), 324–334.
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. doi:10.1016/j.cell.2013.05.039.
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F., & Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389(6648), 251–260. doi:10.1038/38444.
Ma, Y., Jacobs, S. B., Jackson-Grusby, L., Mastrangelo, M. A., Torres-Betancourt, J. A., Jaenisch, R., et al. (2005). DNA CpG hypomethylation induces heterochromatin reorganization involving the histone variant macroH2A. Journal of Cell Science, 118(Pt 8), 1607–1616. doi:10.1242/jcs.02291.
Maeshima, K., Imai, R., Tamura, S., & Nozaki, T. (2014). Chromatin as dynamic 10-nm fibers. Chromosoma, 123(3), 225–237. doi:10.1007/s00412-014-0460-2.
Manuyakorn, A., Paulus, R., Farrell, J., Dawson, N. A., Tze, S., Cheung-Lau, G., et al. (2010). Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: Results from RTOG 9704. Journal of Clinical Oncology, 28(8), 1358–1365. doi:10.1200/JCO.2009.24.5639.
Margueron, R., Justin, N., Ohno, K., Sharpe, M. L., Son, J., Drury, W. J., 3rd, et al. (2009). Role of the polycomb protein EED in the propagation of repressive histone marks. Nature, 461(7265), 762–767. doi:10.1038/nature08398.
Marioni, R. E., Shah, S., McRae, A. F., Chen, B. H., Colicino, E., Harris, S. E., et al. (2015). DNA methylation age of blood predicts all-cause mortality in later life. Genome Biology, 16(1), 25. doi:10.1186/s13059-015-0584-6.
Marsden, M. P., & Laemmli, U. K. (1979). Metaphase chromosome structure: Evidence for a radial loop model. Cell, 17(4), 849–858. doi:10.1016/0092-8674(79)90325-8.
Martin, G. M. (2005). Epigenetic drift in aging identical twins. Proceedings of the National Academy of Sciences of the United States of America, 102(30), 10413–10414. doi:10.1073/pnas.0504743102.
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., & Rogers, J. (2011). Epigenetic mechanisms in Alzheimer’s disease. Neurobiology of Aging, 32(7), 1161–1180. doi:10.1016/j.neurobiolaging.2010.08.017.
McCord, R. P., Nazario-Toole, A., Zhang, H., Chines, P. S., Zhan, Y., Erdos, M. R., et al. (2013). Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Research, 23(2), 260–269. doi:10.1101/gr.138032.112.
McGarvey, K. M., Van Neste, L., Cope, L., Ohm, J. E., Herman, J. G., Van Criekinge, W., et al. (2008). Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Research, 68(14), 5753–5759. doi:10.1158/0008-5472.can-08-0700.
McKenzie, S. J., McLaughlin, D., Clark, J., & Doi, S. A. (2015). The burden of non-adherence to cardiovascular medications among the aging population in Australia: A meta-analysis. Drugs & Aging. doi:10.1007/s40266-015-0245-1.
Medrzycki, M., Zhang, Y., McDonald, J. F., & Fan, Y. (2012). Profiling of linker histone variants in ovarian cancer. Frontiers in Bioscience, 17, 396–406.
Merideth, M. A., Gordon, L. B., Clauss, S., Sachdev, V., Smith, A. C., Perry, M. B., et al. (2008). Phenotype and course of Hutchinson-Gilford progeria syndrome. The New England Journal of Medicine, 358(6), 592–604. doi:10.1056/NEJMoa0706898.
Miquel, P. A. (2014). Aging as alteration. Interdisciplinary Topics in Gerontology, 39, 187–197. doi:10.1159/000358906.
Misteli, T. (2010). Higher-order genome organization in human disease. Cold Spring Harbor Perspectives in Biology, 2(8), a000794. doi:10.1101/cshperspect.a000794.
Mitteldorf, J. (2015). How does the body know how old it is? Introducing the epigenetic clock hypothesis. Interdisciplinary Topics in Gerontology, 40, 49–62. doi:10.1159/000364929.
Morris, M., Iansek, R., Matyas, T., & Summers, J. (1998). Abnormalities in the stride length-cadence relation in Parkinsonian gait. Movement Disorders, 13(1), 61–69. doi:10.1002/mds.870130115.
Muller-Tidow, C., Klein, H. U., Hascher, A., Isken, F., Tickenbrock, L., Thoennissen, N., et al. (2010). Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood, 116(18), 3564–3571. doi:10.1182/blood-2009-09-240978.
Muñoz-Najar, U., & Sedivy, J. M. (2011). Epigenetic control of aging. Antioxidants and Redox Signaling, 14(2), 241–259. doi:10.1089/ars.2010.3250.
Nagarajan, R. P., Zhang, B., Bell, R. J., Johnson, B. E., Olshen, A. B., Sundaram, V., et al. (2014). Recurrent epimutations activate gene body promoters in primary glioblastoma. Genome Research, 24(5), 761–774. doi:10.1101/gr.164707.113.
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell, 113(6), 703–716.
Novikov, L., Park, J. W., Chen, H., Klerman, H., Jalloh, A. S., & Gamble, M. J. (2011). QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Molecular and Cellular Biology, 31(20), 4244–4255. doi:10.1128/MCB.05244-11.
O’Sullivan, R. J., & Karlseder, J. (2012). The great unravelling: Chromatin as a modulator of the aging process. Trends in Biochemical Sciences, 37(11), 466–476. doi:10.1016/j.tibs.2012.08.001.
O’Sullivan, R. J., Kubicek, S., Schreiber, S. L., & Karlseder, J. (2010). Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nature Structural & Molecular Biology, 17(10), 1218–1225. doi:10.1038/nsmb.1897.
Oakes, C. C., Smiraglia, D. J., Plass, C., Trasler, J. M., & Robaire, B. (2003). Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proceedings of the National Academy of Sciences of the United States of America, 100(4), 1775–1780. doi:10.1073/pnas.0437971100.
Obeid, R., & Herrmann, W. (2006). Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Letters, 580(13), 2994–3005. doi:10.1016/j.febslet.2006.04.088.
Obeid, R., Schadt, A., Dillmann, U., Kostopoulos, P., Fassbender, K., & Herrmann, W. (2009). Methylation status and neurodegenerative markers in Parkinson disease. Clinical Chemistry, 55(10), 1852–1860. doi:10.1373/clinchem.2009.125021.
Oberdoerffer, P., & Sinclair, D. A. (2007). The role of nuclear architecture in genomic instability and ageing. Nature Reviews Molecular Cell Biology, 8(9), 692–702. doi:10.1038/nrm2238.
Oh, J. H., Gertych, A., & Tajbakhsh, J. (2013). Nuclear DNA methylation and chromatin condensation phenotypes are distinct between normally proliferating/aging, rapidly growing/immortal, and senescent cells. Oncotarget, 4(3), 474–493.
Palstra, R. J. (2009). Close encounters of the 3C kind: Long-range chromatin interactions and transcriptional regulation. Briefings in Functional Genomics & Proteomics, 8(4), 297–309. doi:10.1093/bfgp/elp016.
Park, Y. S., Jin, M. Y., Kim, Y. J., Yook, J. H., Kim, B. S., & Jang, S. J. (2008). The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Annals of Surgical Oncology, 15(7), 1968–1976. doi:10.1245/s10434-008-9927-9.
Pegoraro, G., Kubben, N., Wickert, U., Gohler, H., Hoffmann, K., & Misteli, T. (2009). Ageing-related chromatin defects through loss of the NURD complex. Nature Cell Biology, 11(10), 1261–1267. doi:10.1038/ncb1971.
Pegoraro, G., & Misteli, T. (2009). The central role of chromatin maintenance in aging. Aging, 1(12), 1017–1022.
Pollex, R. L., & Hegele, R. A. (2004). Hutchinson-Gilford progeria syndrome. Clinical Genetics, 66(5), 375–381. doi:10.1111/j.1399-0004.2004.00315.x.
Popova, E. Y., Grigoryev, S. A., Fan, Y., Skoultchi, A. I., Zhang, S. S., & Barnstable, C. J. (2013). Developmentally regulated linker histone H1c promotes heterochromatin condensation and mediates structural integrity of rod photoreceptors in mouse retina. The Journal of Biological Chemistry, 288(24), 17895–17907. doi:10.1074/jbc.M113.452144.
Prakash, S., Agrawal, S., Cao, J. N., Gupta, S., & Agrawal, A. (2013). Impaired secretion of interferons by dendritic cells from aged subjects to influenza: Role of histone modifications. Age, 35(5), 1785–1797. doi:10.1007/s11357-012-9477-8.
Prieto, E., Hizume, K., Kobori, T., Yoshimura, S. H., & Takeyasu, K. (2012). Core histone charge and linker histone H1 effects on the chromatin structure of Schizosaccharomyces pombe. Bioscience, Biotechnology, and Biochemistry, 76(12), 2261–2266. doi:10.1271/bbb.120548.
Rattner, J. B., Saunders, C., Davie, J. R., & Hamkalo, B. A. (1982). Ultrastructural organization of yeast chromatin. Journal of Cell Biology, 93(1), 217–222.
Richmond, T. J., Finch, J. T., Rushton, B., Rhodes, D., & Klug, A. (1984). Structure of the nucleosome core particle at 7 resolution. Nature, 311(5986), 532–537. doi:10.1038/311532a0.
Riggs, A. D. (1975). X inactivation, differentiation, and DNA methylation. Cytogenetics and Cell Genetics, 14(1), 9–25.
Robertson, K. D. (2005). DNA methylation and human disease. Nature Reviews Genetics, 6(8), 597–610. doi:10.1038/nrg1655.
Robertson, K. D., Uzvolgyi, E., Liang, G., Talmadge, C., Sumegi, J., Gonzales, F. A., et al. (1999). The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Research, 27(11), 2291–2298.
Robinson, P. J., An, W., Routh, A., Martino, F., Chapman, L., Roeder, R. G., et al. (2008). 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. Journal of Molecular Biology, 381(4), 816–825. doi:10.1016/j.jmb.2008.04.050.
Robinson, P. J., Fairall, L., Huynh, V. A., & Rhodes, D. (2006). EM measurements define the dimensions of the “30-nm” chromatin fiber: Evidence for a compact, interdigitated structure. Proceedings of the National Academy of Sciences of the United States of America, 103(17), 6506–6511. doi:10.1073/pnas.0601212103.
Robinson, P. J., & Rhodes, D. (2006). Structure of the ‘30 nm’ chromatin fibre: A key role for the linker histone. Current Opinion in Structural Biology, 16(3), 336–343. doi:10.1016/j.sbi.2006.05.007.
Rodrigues, H. F., Souza, T. A., Ghiraldini, F. G., Mello, M. L., & Moraes, A. S. (2014). Increased age is associated with epigenetic and structural changes in chromatin from neuronal nuclei. Journal of Cellular Biochemistry, 115(4), 659–665. doi:10.1002/jcb.24705.
Rodriguez-Paredes, M., & Esteller, M. (2011). Cancer epigenetics reaches mainstream oncology. Nature Medicine, 17(3), 330–339. doi:10.1038/nm.2305.
Rogakou, E. P., & Sekeri-Pataryas, K. E. (1999). Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Experimental Gerontology, 34(6), 741–754.
Rogenhofer, S., Kahl, P., Mertens, C., Hauser, S., Hartmann, W., Buttner, R., et al. (2012). Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU International, 109(3), 459–465. doi:10.1111/j.1464-410X.2011.10278.x.
Rolf, M. K. (2015). Addressing the needs of our aging population through managed long-term care. Home Healthcare Now, 33(3), 177–178. doi:10.1097/nhh.0000000000000201.
Routh, A., Sandin, S., & Rhodes, D. (2008). Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proceedings of the National Academy of Sciences of the United States of America, 105(26), 8872–8877. doi:10.1073/pnas.0802336105.
Ryan, J. M., & Cristofalo, V. J. (1972). Histone acetylation during aging of human cells in culture. Biochemical and Biophysical Research Communications, 48(4), 735–742.
Ryba, T., Hiratani, I., Lu, J., Itoh, M., Kulik, M., Zhang, J., et al. (2010). Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Research, 20(6), 761–770. doi:10.1101/gr.099655.109.
Sanyal, A., Bau, D., Marti-Renom, M. A., & Dekker, J. (2011). Chromatin globules: A common motif of higher order chromosome structure? Current Opinion in Cell Biology, 23(3), 325–331. doi:10.1016/j.ceb.2011.03.009.
Scaffidi, P., & Misteli, T. (2006). Lamin A-dependent nuclear defects in human aging. Science, 312(5776), 1059–1063. doi:10.1126/science.1127168.
Schreiber, K. H., & Kennedy, B. K. (2013). When lamins go bad: Nuclear structure and disease. Cell, 152(6), 1365–1375. doi:10.1016/j.cell.2013.02.015.
Sedelnikova, O. A., & Bonner, W. M. (2006). GammaH2AX in cancer cells: A potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle, 5(24), 2909–2913.
Sedivy, J. M., Banumathy, G., & Adams, P. D. (2008). Aging by epigenetics - A consequence of chromatin damage? Experimental Cell Research, 314(9), 1909–1917. doi:10.1016/j.yexcr.2008.02.023.
Seeber, A., Dion, V., & Gasser, S. M. (2014). Remodelers move chromatin in response to DNA damage. Cell Cycle, 13(6), 877–878. doi:10.4161/cc.28200.
Seligson, D. B., Horvath, S., McBrian, M. A., Mah, V., Yu, H., Tze, S., et al. (2009). Global levels of histone modifications predict prognosis in different cancers. The American Journal of Pathology, 174(5), 1619–1628. doi:10.2353/ajpath.2009.080874.
Sharman, E. H. (2010). Sirtuins and mammalian aging. In S. Bondy & K. Maiese (Eds.), Aging and age-related disorders oxidative stress in applied basic research and clinical practice. New York: Humana.
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119(7), 941–953. doi:10.1016/j.cell.2004.12.012.
Shin, D. M., Kucia, M., & Ratajczak, M. Z. (2011). Nuclear and chromatin reorganization during cell senescence and aging - a mini-review. Gerontology, 57(1), 76–84. doi:10.1159/000281882.
Shumaker, D. K., Dechat, T., Kohlmaier, A., Adam, S. A., Bozovsky, M. R., Erdos, M. R., et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proceedings of the National Academy of Sciences of the United States of America, 103(23), 8703–8708. doi:10.1073/pnas.0602569103.
Singhal, R. P., Mays-Hoopes, L. L., & Eichhorn, G. L. (1987). DNA methylation in aging of mice. Mechanisms of Ageing and Development, 41(3), 199–210.
So, K., Tamura, G., Honda, T., Homma, N., Endoh, M., Togawa, N., et al. (2006a). Quantitative assessment of RUNX3 methylation in neoplastic and non-neoplastic gastric epithelia using a DNA microarray. Pathology International, 56(10), 571–575. doi:10.1111/j.1440-1827.2006.02010.x.
So, K., Tamura, G., Honda, T., Homma, N., Waki, T., Togawa, N., et al. (2006b). Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Science, 97(11), 1155–1158. doi:10.1111/j.1349-7006.2006.00302.x.
Solovei, I., Kreysing, M., Lanctot, C., Kosem, S., Peichl, L., Cremer, T., et al. (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell, 137(2), 356–368. doi:10.1016/j.cell.2009.01.052.
Song, J. S., Kim, Y. S., Kim, D. K., Park, S. I., & Jang, S. J. (2012). Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathology International, 62(3), 182–190. doi:10.1111/j.1440-1827.2011.02776.x.
Soutoglou, E., & Misteli, T. (2007). Mobility and immobility of chromatin in transcription and genome stability. Current Opinion in Genetics & Development, 17(5), 435–442. doi:10.1016/j.gde.2007.08.004.
Sporn, J. C., Kustatscher, G., Hothorn, T., Collado, M., Serrano, M., Muley, T., et al. (2009). Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene, 28(38), 3423–3428. doi:10.1038/onc.2009.26.
Suzuki, Y., & Craigie, R. (2007). The road to chromatin - nuclear entry of retroviruses. Nature Reviews Microbiology, 5(3), 187–196. doi:10.1038/nrmicro1579.
Tark-Dame, M., van Driel, R., & Heermann, D. W. (2011). Chromatin folding – from biology to polymer models and back. Journal of Cell Science, 124(Pt 6), 839–845. doi:10.1242/jcs.077628.
Tempera, I., & Lieberman, P. M. (2014). Epigenetic regulation of EBV persistence and oncogenesis. Seminars in Cancer Biology, 26, 22–29. doi:10.1016/j.semcancer.2014.01.003.
Thoma, F., & Koller, T. (1977). Influence of histone H1 on chromatin structure. Cell, 12(1), 101–107.
Thoma, F., Koller, T., & Klug, A. (1979). Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. The Journal of Cell Biology, 83(2 Pt 1), 403–427.
Tohgi, H., Utsugisawa, K., Nagane, Y., Yoshimura, M., Genda, Y., & Ukitsu, M. (1999). Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Research. Molecular Brain Research, 70(2), 288–292.
Upadhya, B., Taffet, G. E., Cheng, C. P., & Kitzman, D. W. (2015). Heart failure with preserved ejection fraction in the elderly: Scope of the problem. Journal of Molecular and Cellular Cardiology. doi:10.1016/j.yjmcc.2015.02.025.
Uzunova, K., Georgieva, M., & Miloshev, G. (2013). Saccharomyces cerevisiae linker histone-Hho1p maintains chromatin loop organization during ageing. Oxidative Medicine and Cellular Longevity, 2013, 437146. doi:10.1155/2013/437146.
Van Den Broeck, A., Brambilla, E., Moro-Sibilot, D., Lantuejoul, S., Brambilla, C., Eymin, B., et al. (2008). Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clinical Cancer Research, 14(22), 7237–7245. doi:10.1158/1078-0432.CCR-08-0869.
van Leeuwen, H., Okuwaki, M., Hong, R., Chakravarti, D., Nagata, K., & O’Hare, P. (2003). Herpes simplex virus type 1 tegument protein VP22 interacts with TAF-I proteins and inhibits nucleosome assembly but not regulation of histone acetylation by INHAT. The Journal of General Virology, 84(Pt 9), 2501–2510.
Verreault, A., Kaufman, P. D., Kobayashi, R., & Stillman, B. (1996). Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell, 87(1), 95–104.
Waki, T., Tamura, G., Sato, M., & Motoyama, T. (2003). Age-related methylation of tumor suppressor and tumor-related genes: An analysis of autopsy samples. Oncogene, 22(26), 4128–4133. doi:10.1038/sj.onc.1206651.
Wang, G. P., Ciuffi, A., Leipzig, J., Berry, C. C., & Bushman, F. D. (2007). HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Research, 17(8), 1186–1194. doi:10.1101/gr.6286907.
Wang, Y., Wysocka, J., Sayegh, J., Lee, Y. H., Perlin, J. R., Leonelli, L., et al. (2004). Human PAD4 regulates histone arginine methylation levels via demethylimination. Science, 306(5694), 279–283. doi:10.1126/science.1101400.
Wang, R. H., Zheng, Y., Kim, H. S., Xu, X., Cao, L., Luhasen, T., et al. (2008). Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Molecular Cell, 32(1), 11–20. doi:10.1016/j.molcel.2008.09.011.
Weintraub, D., Comella, C. L., & Horn, S. (2008). Parkinson’s disease–Part 1: Pathophysiology, symptoms, burden, diagnosis, and assessment. The American Journal of Managed Care, 14(2 Suppl), S40–48.
Widom, J., & Klug, A. (1985). Structure of the 300A chromatin filament: X-ray diffraction from oriented samples. Cell, 43(1), 207–213.
Wilson, V. L., Smith, R. A., Ma, S., & Cutler, R. G. (1987). Genomic 5-methyldeoxycytidine decreases with age. The Journal of Biological Chemistry, 262(21), 9948–9951.
Wolffe, A. P. (1998). Epigenetics. Introduction. Novartis Foundation Symposium, 214, 1–5.
Wolffe, A. P., & Matzke, M. A. (1999). Epigenetics: Regulation through repression. Science, 286(5439), 481–486.
Wood, J. G., & Helfand, S. L. (2013). Chromatin structure and transposable elements in organismal aging. Frontiers in Genetics, 4, 274. doi:10.3389/fgene.2013.00274.
Woodcock, C. L. (2006). Chromatin architecture. Current Opinion in Structural Biology, 16(2), 213–220. doi:10.1016/j.sbi.2006.02.005.
Yanagawa, N., Tamura, G., Oizumi, H., Kanauchi, N., Endoh, M., Sadahiro, M., et al. (2007). Promoter hypermethylation of RASSF1A and RUNX3 genes as an independent prognostic prediction marker in surgically resected non-small cell lung cancers. Lung Cancer, 58(1), 131–138. doi:10.1016/j.lungcan.2007.05.011.
Yuan, H., Su, L., & Chen, W. Y. (2013). The emerging and diverse roles of sirtuins in cancer: A clinical perspective. OncoTargets and Therapy, 6, 1399–1416. doi:10.2147/OTT.S37750.
Zane, L., Sharma, V., & Misteli, T. (2014). Common features of chromatin in aging and cancer: Cause or coincidence? Trends in Cell Biology. doi:10.1016/j.tcb.2014.07.001.
Zhang, K., Chen, Y., Zhang, Z., & Zhao, Y. (2009a). Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. Journal of Proteome Research, 8(2), 900–906. doi:10.1021/pr8005155.
Zhang, Y., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Bird, A., & Reinberg, D. (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes & Development, 13(15), 1924–1935.
Zhang, R., Poustovoitov, M. V., Ye, X., Santos, H. A., Chen, W., Daganzo, S. M., et al. (2005). Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Developmental Cell, 8(1), 19–30. doi:10.1016/j.devcel.2004.10.019.
Zhang, Y., Zhang, M., Dong, H., Yong, S., Li, X., Olashaw, N., et al. (2009b). Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene, 28(3), 445–460. doi:10.1038/onc.2008.388.
Zhang, L., Zhong, K., Dai, Y., & Zhou, H. (2009c). Genome-wide analysis of histone H3 lysine 27 trimethylation by ChIP-chip in gastric cancer patients. Journal of Gastroenterology, 44(4), 305–312. doi:10.1007/s00535-009-0027-9.
Acknowledgements
The authors are thankful to Djulia Milcheva and Mathew Serkedjiev for the fruitful discussions and comments on the section regarding age-associated diseases. The authors highly acknowledge Toni Efremov for all technical assistance during the preparation of the current book chapter.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Abbreviations
Abbreviations
- 4C:
-
Circularized Chromosome Conformation Capture
- 5C:
-
Carbon-Copy Chromosome Conformation Capture
- AFM :
-
Atomic force microscope
- bp:
-
Base pairs
- CGIs:
-
CpG islands
- ChCA:
-
Chromatin Comet Assay
- ChIA-PET:
-
Chromatin Interaction Analysis with Paired-End Tag Sequencing
- DNA:
-
Deoxyribonucleic acid
- DNMT:
-
DNA methyltransferase
- FISH:
-
Fluorescence In Situ Hybridization
- H1:
-
Linker histone
- HGPS:
-
Hutchinson-Gilford progeria syndrome
- Hi-C:
-
Genome-wide chromosome conformation capture
- HMGA:
-
High Mobility Group protein A
- HMT:
-
Histone Methyltransferase
- HP1:
-
Heterochromatin Protein 1
- Kb:
-
Kilobases
- Mb:
-
Megabases
- Nm:
-
Nanometers
- ROS:
-
Reactive Oxygen Species
- SAHF:
-
Senescence -Associated Heterochromatin Foci
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Georgieva, M., Staneva, D., Miloshev, G. (2016). Epigenetic Significance of Chromatin Organization During Cellular Aging and Organismal Lifespan. In: Hollar, D. (eds) Epigenetics, the Environment, and Children’s Health Across Lifespans. Springer, Cham. https://doi.org/10.1007/978-3-319-25325-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-25325-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-25323-7
Online ISBN: 978-3-319-25325-1
eBook Packages: MedicineMedicine (R0)