
Abstract
Key message
Sustaining yield gains of grain legume crops under growing salt-stressed conditions demands a thorough understanding of plant salinity response and more efficient breeding techniques that effectively integrate modern omics knowledge.
Abstract
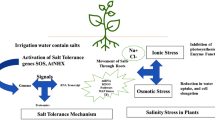
Grain legume crops are important to global food security being an affordable source of dietary protein and essential mineral nutrients to human population, especially in the developing countries. The global productivity of grain legume crops is severely challenged by the salinity stress particularly in the face of changing climates coupled with injudicious use of irrigation water and improper agricultural land management. Plants adapt to sustain under salinity-challenged conditions through evoking complex molecular mechanisms. Elucidating the underlying complex mechanisms remains pivotal to our knowledge about plant salinity response. Improving salinity tolerance of plants demand enriching cultivated gene pool of grain legume crops through capitalizing on ‘adaptive traits’ that contribute to salinity stress tolerance. Here, we review the current progress in understanding the genetic makeup of salinity tolerance and highlight the role of germplasm resources and omics advances in improving salt tolerance of grain legumes. In parallel, scope of next generation phenotyping platforms that efficiently bridge the phenotyping–genotyping gap and latest research advances including epigenetics is also discussed in context to salt stress tolerance. Breeding salt-tolerant cultivars of grain legumes will require an integrated “omics-assisted” approach enabling accelerated improvement of salt-tolerance traits in crop breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
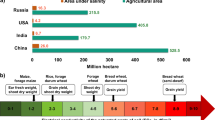
Salt stress causes considerable loss in agricultural production worldwide through severely impacting upon plant growth (Amitai et al. 1995; Banzai et al. 2002). Salinity-affected soils are prevalent in arable and irrigated lands in arid and semi-arid climates with considerable higher evapotranspiration (Shanon 1986; Sharifia et al. 2007; Manchanda and Sharma 2008; Li et al. 2014; Kaashyap et al. 2017). Salinity remains one of the key drivers that contribute to soil toxicity in tropical Asia (Greenland 1984). The major factors that aggravate the challenge of soil salinity stress include low precipitation, high surface evaporation, depletion of ground water and inappropriate agricultural practices including improper ‘drainage in irrigated land’ (Jamil et al. 2011; Munns and Gilliham 2015). Wang et al. (2003) highlighted the growing expansion of salt-stressed area, with authors predicting nearly 30% loss in land due to salinity stress within next 25 years. In China, 9.2 mha area representing 6.62% of the total cultivated land is critically challenged by salinity stress (Yang et al. 2008). The global acreage constrained by salt stress includes a total of 45 mha irrigated and 32 mha hardy lands (Munns and Tester 2008; FAO 2015).
Grain legumes are important in relation to global food security (Zhu et al. 2005; Bohra et al. 2015), and their significance is attributable to higher contents of protein and other mineral nutrients (Broughton et al. 2003; Bohra et al. 2014; Foyer et al. 2016; Considine et al. 2017). Their ability to fix atmospheric nitrogen helps improving soil fertility and also contributes to sustainability of cereal–legume based cropping systems (Foyer et al. 2016). Like cereals, production of grain legume crops is also severely affected by salinity stress worldwide. Therefore, improved cultivars with higher salt tolerance are required to maintain their yield potential under salt-stressed condition (Kaashyap et al. 2017). Here, we discuss the impact of salinity stress on five major grain legumes viz. chickpea, common bean, cowpea, field pea and soybean together with describing the adaptive mechanisms against salinity stress. This is followed by a brief account on current knowledge about genetic resources, and genetics and genomics of salinity tolerance in these crops. Finally, we outline the scope of bridging the existing phenotype–genotype gap in light of the next generation plant phenotyping techniques.
Effects of salinity stress on grain legumes
Plants stressed with salinity show pertuberations in cellular metabolism and plant growth and development due to impaired CO2 assimilation (Bayuelo-Jiménez et al. 2002a; Chen and Yu 2007; Khan et al. 2015, 2017), hindrance in nutrient uptake (Ahmed and Jhon 2005; Gama et al. 2007; Shahid et al. 2012), defective cytosolic enzymes, osmotic stress, ion toxicity (excess Na+/Cl−), hormonal dysfunction, oxidative stress (Essa 2002; Hernandez and; Almansa 2002; Bayuelo-Jiménes et al. 2003; Ahmad and Jhon 2005; Najafi et al. 2006; Shahid et al. 2012a, b), ultimately leading to cell death (Shabala 2009). Comprehensive reviews detailing impacts of salinity stress on grain legumes are made elsewhere (Manchanda and Garg 2008; Flower et al. 2010; Farooq et al. 2017).
Adaptive mechanisms conferring salinity tolerance in grain legume crops
Plants respond to salinity stress through evoking a range of physiological, biochemical, and molecular mechanisms (Ashraf and Harris 2004; Munns and Tester 2008; Gupta and Huang 2014; Roy et al. 2014; Acosta-Motos et al. 2017; Liang et al. 2018), which can be categorized into three major classes (i) ‘ion exclusion’ to eliminate Na+ and Cl− ions from roots when their accumulation becomes toxic, (ii) ‘tissue tolerance’ allowing ‘compartmentalized of toxic ions at cellular and intracellular level’ (Roy et al. 2014) and (iii) ‘osmotic tolerance’(Parida et al. 2005; Munns and Tester 2008; Roy et al. 2014; Deinlein et al. 2014; Ismail and Horie 2017; Negrao et al. 2017). Mechanisms relying on ion exclusion and tissue tolerance against salinity have been reported in soybean (Durand and Lacan 1994; Umezawa et al. 2000; Tsai 2003; Lee et al. 2004; Yu et al. 2005; Li et al. 2006; Sun et al. 2006; Wong et al. 2013; Guan et al. 2014a; Qi et al. 2014; Do et al. 2016; Liu et al. 2016; Patil et al. 2016) and in pea (Pandolfi et al. 2012). Salinity tolerance resulting from ‘osmotic tolerance’ mechanism has been reported in soybean (Phang et al. 2008; Ozge and Atak 2012; Wu et al. 2014), pea (Shahid et al. 2012), chickpea (Singh 2004) and in common bean (Chen et al. 2009). The physiological and biochemical processes leading to salinity tolerance are intensively investigated in model as well as industrial crops like soybean; however, the underlying mechanisms and pathways remain to be elucidated in the case of other grain legumes.
Grain legume germplasm resources for improving salinity stress tolerance
Characterisation and utilization of genetic resources is key to genetic improvement of any crop. Significant progress has been achieved in crops for breeding salinity tolerance, notably in cereals like rice and wheat [for details see Ashraf and Wu (2011), Ismail and Horie (2017)]. However, relatively narrow genetic base of breeding programs of grain legume crops has hampered the progress of breeding against salinity (Sharma et al. 2017).
In recent years, efforts were made to explore the genetic vatiation for salinity tolerance in different legume crops. Chickpea genotypes show a wide range of variation in their response to salinity stress (Lauter and Munns 1986; Maliro et al. 2004; Serraj et al. 2004; Vadez et al. 2007; Krishnamurthy et al. 2011; Turner et al. 2013), and the variation in the level of salinity tolerance is reported across different growth stages. For instance, genotypes such as C 10, C 14, C 16, C 17, C 19, C 28 and C 29 could tolerate salinity up to 6 dSm−1 at germination and seedling stage (Al-Mutawa 2003). The genotype L 550 showed tolerance under saline conditions on account of its ability to tolerate Na+ toxicity (Lauter and Munns 1986). Similarly, two genotypes ICCC 32 and ICCL 86446 were reported to show higher tolerance against Cl− ion toxicity (Dua 1992). Higher tolerance of kabuli chickpea than the desi types was demonstrated through analysis of 211 minicore collection (Serraj et al. 2004). By contrast, Vadez et al. (2007) found desi chickpea to be more tolerant than the kabuli types following evaluation of 263 germplasm lines under both salinity and controlled conditions. Yield under stressed conditions remains an important parameter for assessing stress tolerance of plants, and significant genetic variation has been reported for yield parameters in both desi and kabuli chickpea (Dua and Sharma 1995; Vadez et al. 2007, 2012; Turner et al. 2013). Turner et al. (2013) recorded upto 27-fold differences among 55 chickpea genotypes for seed yield at 40 mM NaCl. Given the considerable impact of genotype × environment (G × E) interaction on plant salinity tolerance, multi-location testing has enabled identification of the salinity-tolerant chickpea such as ICC 9942 (Vadez et al. 2007; Krishnamurthy et al. 2011; Turner et al. 2013).
In common bean, analysis of 132 wild and 11 cultivated accessions using parameters like susceptibility index, root and shoot ratio demonstrated their differential response to salinity (Bayuelo-Jiménes et al. 2002). Various research groups have found significant variation for salinity tolerance among wild common beans (Bayuelo-Jiménes et al. 2002a, b; Bayuelo-Jiménes et al. 2003; Goerzt and Coons 1991). For example, higher germination of Phaseolus species like P. filiformis, P. angustissimus, P. leptostachyus, and P. microcarpus observed at 120 mM NaCl underscores the importance of wild species vis a vis pre-breeding programs (Bayuelo-Jiménes et al. 2002a). Based on the survival rate, Phaseolus genotype HRS 516 showed tolerance to varying NaCl concentrations such as 50 mM and 100 mM (Gama et al. 2007).
Analysis of 25 cowpea genotypes at 85 and 170 nmol NaCl L−1 for germination percentage facilitated identification of the genotype CB 27 as the most salinity tolerant and others including CB 88, CB 3, CB 5, Tardon, Cuarenteno and CB 46 as moderately tolerant (Murillo-Amador et al. 2001). By examining Na+ accumulation in root and shoot, and biomass production at varying NaCl concentrations, i.e., 0, 85, and 170 mM, four local cowpea accessions viz. Pacen˜o’, Tardon, Sonorense and Cuarenten˜o and three accessions from California CB 46, CB 27 and CB 3 showed tolerance (Murillo-Amador et al. 2006). Interestingly, higher accumulation of Na+ was recorded in roots in comparison to shoot in salt-tolerant genotypes. As shown in Table 1, greater tolerance of the cowpea genotypes 210856, 211557 and Asebot against salinity was evident based on root and shoot vigor under salinity stress (Gogile et al. 2013).
In pea, variation for traits such as plant height, plant growth rate, roots and shoot biomass was revealed following screening of 780 pea accessions under salinity stress (Leonforte et al. 2013a, b). Similarly, another study involving 30 pea genotypes reported variation for germination percentage, root and shoot weight and inorganic osmolytes (Shahid et al. 2012). The salt tolerance of the genotypes Samarina Zard, Climax, 9800-5 was found to be due to less accumulation of toxic Na+ in leaf and a higher Na+/K+ ratio, and abundant antioxidant enzymatic activities and osmolyte content under salinity stress (Shahid et al. 2012a, b).
In soybean, salinity tolerance traits exhibit a wide range of variation (Shao et al. 1986; Wang and Shannon 1999) in both cultivated (Glycine max) and wild relatives (G. soja, G. tomentella and G. argyrea) (Lenis et al. 2011). Tolerant soybean exhibited limited leaf scorching and retained higher chlorophyll content under salinity stress. Wide spectrum of salinity tolerance was revealed in soybean encompassing various growth stages through analyzing a large collection of 1716 germplasm lines, representative of diversity in China provinces (Shao et al. 1986). The authors found seven genotypes showing tolerant reactions at all developmental stages. Later, the same group obtained eighty tolerant genotypes after screening more than 10,000 soybean lines under salinity stress (Shao et al. 1993). Similarly, significant variation for leaf Na+ accumulation and biomass reduction was reported in three wild soybean species viz. G. soja, G. tomentella and G. tabacina under salinity stress (Kao et al. 2006). Perennial soybean displayed tolerance to NaCl up to 17.5 g L−1 in comparison to G. max (5.2–8.0 g L−1), with former showing lower leaf chlorosis (Petalone et al. 1997). Higher level of salinity tolerance has been reported in BB 52 population derived from wild G. soja (Wu and Yu 2009). Examination of salinity tolerance of BB 52, cultivar N 23674 and their hybrid (BB 52 × N 23674) allowed authors to propose restricting Cl− to plant leaves as the possible mechanism imparting salt tolerance to soybean (Zhang et al. 2011). Likewise, the genotype BB 52 was reported as a promising source of salinity tolerance given the abundance of anti-oxidant enzymatic (Chen et al. 2013) and other physiological activities viz., higher relative water content (RWC) under salt stress (Wu et al. 2014). Additionally, contrasting mechanisms of salinity tolerance between G. max (preventing excess Cl− ions to leaf and shoot) and G. soja (prevention of excess Na+ ions from root to leaf and stem) offer greater possibilities of broadening the genetic basis through interspecific hybridization (Luo et al. 2005). These studies highlight the significance of wild and perennial soybean to improving salinity tolerance of cultivated soybean.
Crop wild relatives (CWRs) for harnessing novel variation for salinity tolerance traits
The bottleneck effects associated with crop domestication followed by intensive selection of high yielding lines have caused serious loss of genetic diversity in current crop breeding programs (Tanksley and McCouch 1997). The considerable loss in genetic diversity of food crops is evident at farmers’s field also (Massawe et al. 2016). Therefore, novel allelic diversity must be introduced in breeding programmes to exploit genetic variations related to various resilience traits such as tolerance to salinity stress (Brozynska et al. 2016). Though the CWRs of grain legumes remain underutilized (Sharma 2017), efforts have been made in recent years to harness the variation for the traits relevant to salinity tolerance (Bayuelo-Jiménes et al. 2002a, b; Kao et al. 2006; Maliro et al. 2008; Wu and Yu 2009; Lenis et al. 2011). Recent genome sequencing attempts have allowed decoding whole genomes of CWRs, thus shedding new light on novel allele(s)/genomic information pertaining to salinity tolerance (Guan et al. 2014b; Qi et al. 2014; Brozynska et al. 2016; Munoz et al. 2017). Improving crop performance under increasing salinity stress warrants greater utilization of CRWs in breeding programs to allow untapped genetic variation flowing from CWRs to elite agronomic bases (Wang et al. 2017).
Genetics of salt tolerance and efforts of conventional breeding for salinity stress breeding
Greater understanding of the genetic basis of traits having relevance to salinity stress is important for improving salinity tolerance in crops (Lee et al. 2009; Arzani and Ashraf 2016). Breeding for salinity tolerant crop varieties is time consuming given the multi-genic inheritance and ‘multi-component nature’ of salinity stress tolerance (DeRose-Wilson and Gaut 2011; Cabot et al. 2014; Negrao et al. 2017). Hence, direct selection for higher yield as a measure of salinity tolerance cannot be deemed very suitable (Ashraf 2004). Complexities in both genetic and physiological mechanisms of salinity tolerance in crop plants have been thoroughly discussed elsewhere (for details see Flowers 2004).
Classical genetics and conventional breeding approaches relying on phenotypic variation were implemented in grain legumes to understand salinity stress (Abel 1969; Dua and Sharma 1995; Serraj et al. 2004; Maliro et al. 2008; Xu and Tuyen 2010). Majority of these genetic analyses on salt tolerance are confined to only model legumes such as soybean. Examples include a classical genetics study by Abel (1969) that suggested a single dominant gene Ncl controlling salinity tolerance in soybean. This observation concorded with that of Shao et al. (1994) who proposed a single dominant gene for salt tolerance in soybean. Subsequently, a new allele from PI 483463, designated as Ncl2 (Lee et al. 2009), was found to be different from the salinity tolerant gene reported from G. max line S100. Earlier, Luo et al. (2004) reported polygenic inheritance of salt tolerance in soybean based on the inheritance patterns inferred from crosses (Nannong 88–31 × Jackson and Nannong 1138–2 × Nannong 88–31). A list of genes contributing to salinity tolerance in soybean is given in Table 2. In chickpea, a diallel cross analysis revealed presence of both additive and dominance gene effects with higher dominance effects for the three traits (seed yield, pods per plant and seeds per plant) measured under salinity stress (Asraf and Waheed 1998). Similarly, generation mean analysis (GMA) in chickpea involving crosses derived from ICC 6263 (salt sensitive) × ICC 1431 (salt tolerant) for yield related traits (pods per plant, seeds per plant and seed yield) showed significant dominant effects under controlled condition, whereas additive effects were significant for the given yield traits under salinity stress (Samineni et al. 2011). Authors’ proposition of preponderance of additive effects for yield related traits under salinity stress, however, could not gather support from previous studies (Asraf and Waheed 1998). Availability of advanced mating designs such as nested association mapping (NAM), multi-parent advanced generation inter-cross (MAGIC) could play a larger role in resolving the complex genetic make up of such traits (Pandey et al. 2016).
Discovery of QTLs/candidate genes controlling salt tolerance in grain legumes
Current advances in genotyping technologies have allowed discovery and assay of genome-wide genetic markers to locate QTL(s) controlling salinity tolerance in various legume crops (Vadez et al. 2012; Leonforte et al. 2013; Guan et al. 2014a, b; Qi et al. 2014; Pushpavalli et al. 2015; Do et al. 2016; Liu et al. 2016).
Conventional QTL mapping
QTL mapping is an important technique to dissect the genetic architecture of complex traits like salinity stress that are governed by a variety of gene(s)/QTLs. In chickpea, analysis of recombinant inbreds (JG 62 × ICCV 2) enabled identification of one major QTL on LG03 governing 19% phenotypic variation (PV) for high seed yield under salinity condition (Vadez et al. 2012). Additionally, QTLs associated with seed number, pod number and 100-seed weight under both saline and non-saline conditions were mapped on LG06. Recently, two major QTLs controlling salinity tolerance were detected in chickpea on LGs05 and 07 from the population ICCV 2 × JG 11 (Pushpavalli et al. 2015). In parallel, the authors suggested a set of 48 putative candidate genes within the QTL-containing region, which encode various proteins including ion transport, ABA biosynthesis, and transcription factors (TFs). Table 3 enlists various QTLs associated with salinity tolerance-related traits in different grain legumes.
In soybean, one major QTL conferring salt tolerance was reported on LG (N) from an intra-specific population S100 × Tokyo (Lee et al. 2004). Subsequent analyses based on inter-specific (Jackson × JWS 156-1) and intra-specific (FT-Abyara × C 01) (Jin dou No. 6 × 0197) (Hamweih and Xu 2008; Hamweih et al. 2011) populations further supported presence of salt tolerance associated QTL on LGN. Chen et al. (2008) reported a total of eight putative salt tolerance QTLs explaining upto 19.7% PV. The QTLs viz. qpsdG.1 (obtained from green house condition) and the qtrG.1 (detected under field condition) detected on the same location on LG (G) flanked by the markers Sat_164 and Sat_358. Additionally, the QTL qppsN.1 co-localized with same genomic region on LG (N) as suggested earlier by Lee and colleagues.
In soybean, Ha et al. (2013) found one major QTL for salinity tolerance on chromosome 3 by analyzing a RIL population (PI 483463 × Hutcheson) with simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers. This QTL region corresponded with the genomic region suggested earlier by Lee et al. (2004) as associated with salinity tolerance in soybean. Considering tolerance at germination stage, 11 QTLs (related to germination indices) contributing to salt tolerance were mapped in a population (Kefeng1 × Nannong1138-2) (Kan et al. 2016), with the QTL-containing regions showing agreement with previous reports in soybean (Lee et al. 2004; Chen et al. 2008; Hamwieh and Xu 2008; Hamwieh et al. 2011; Ha et al. 2013; Kan et al. 2015).
In pea, two QTLs Salt index_QTL 1(flanked by SNP_100000313 and SNP_100000353) and Salt index_QTL 2 (flanked by SNP_100000318 and SNP_100000130) were reported on LGs Ps III and Ps VII, respectively (Leonforte et al. 2013). The authors also identified a candidate gene Medtr3g073300.1 that codes for a protein associated with salt tolerance. An updated list of QTLs pertaining to salinity tolerance in various crops is available at PLANTSTRESS site (http://www.plantstress.com/biotech/index.asp?Flag=1).
Highly saturated genetic linkage maps enabled by high-density genotyping assays allow better prioritization of candidate gene(s) for downstream analyses and finally, pinpointing the causative locus and its cloning. A major effect QTL for alkaline salt tolerance was fine mapped to a 3.33-cM region on LG17 in soybean (Tuyen et al. 2013), and further analysis of this genomic region led to the identification of four putative candidate genes Glyma17g15000, Glyma17g15150, Glyma17g15520, and Glyma17g15580 (Tuyen et al. 2013). Similarly, Ha et al. (2013) narrowed down a major QTL on LG03 to a 658-kb region harbouring two candidate genes viz. Glyma03g32890 and Glyma03g32900. Interestingly, different research groups have confirmed the presence of salt tolerance-controlling gene (GmCHX1/GmSALT3/Ncl) on chromosome 3 (Guan et al. 2014a, b; Qi et al. 2014; Do et al. 2016; Liu et al. 2016). By further narrowing down the GmCHX1 locus to a 388-Kb region, Qi et al. (2014) identified two candidate genes Glyma03g32890 and Glyma03g32900 having analogy to cation H+ exchanger (CHX) gene. The salt tolerance of W05 was attributed to the absence of a retrotransposon element in ion transporter gene GmCHX1as compared to the salt sensitive genotypes C08 and William 82 having retrotransposon inserted within the gene. Similarly, a 17.5-kb region (GmSALT3 locus) on chromosome 3 harbours a candidate gene Glyma03g32900.1 coding for ‘cation/H+ exchanger family and contributes to salt tolerance via reducing shoot accumulation of Na+ in soybean (Guan et al. 2014a). A single dominant gene for salinity tolerance was mapped on chromosome 3 within a 209-kb region (Hamwieh and Xu2008), and the observation showed agreement with earlier report of Lee et al. (2004). Later, the Ncl locus was assigned to a 16.6-kb region and enabled delineation of the candidate gene Glyma03g32900 (Do et al. 2016). Unlike the major grain legume crops discussed above, limited progress has been achieved towards identification of salinity tolerant QTL(s) in lesser-studied legumes like cowpea. Nevertheless, improving capacities of genotyping and phenotyping technologies could further enable high-resolution trait mapping and cloning of QTLs responsible for salinity tolerance in grain legumes.
Genome wide association studies (GWAS)
Association mapping/GWAS is receiving greater attention due to its ability to improve the resolution of the QTL detection without investing extra efforts on population development (Bohra 2013). Higher precision and allelic richness of GWAS has permitted access to functional genetic variants for salt tolerance traits in genetically diverse germplasms of grain legumes (Guan et al. 2014b; Qi et al. 2014; Kan et al. 2015, 2016; Moghaddam et al. 2016; Patil et al. 2016; Zhang et al. 2016; Hoyos-Villegas et al. 2017; Ravelombola et al. 2017; Xu et al. 2017). For instance, SSR-based association analysis of 196 soybean landraces revealed four significant marker trait associations (SMTAs) for salt tolerance imbibition rate (ST-IR) trait on LGs06 and 07, five SMTAs for salt tolerance germination index (ST-GI) on LGs 07 and 19, and eight SMTAs for ST-GR traits on LGs 01, 02, 03, 06, 07, and 19 (Kan et al. 2016). Interestingly, the reported SMTAs overlapped with the genomic regions reported earlier for having association with salinity tolerance in soybean (Lee et al. 2004; Hamwieh and Xu 2008; Ha et al. 2013; Guan et al. 2014b; Zhang et al. 2014; Kan et al. 2015). Likewise, Kan et al. (2015) detected one SMTA for salt tolerant index at germination stage through GWAS of 191 soybean landraces with 1,142 SNPs, thus offering a set of nine candidate genes. Similarly, GWAS of 106 soybean lines with SoySNP50Kchip (Song et al. 2013) revealed SMTAs on chromosome 3 for physiological parameters like leaf chlorophyll content and leaf scorch ratio (Patil et al. 2016). Importantly, this genomic region overlapped with the GmCHX1 locus known to control salinity tolerance in soybean (Guan et al. 2014a; Qi et al. 2014). Another GWA study on 283 soybean lines using SoySNP50K chip revealed nine genomic regions showing significant association with leaf chloride concentration and leaf chlorophyll content (Zeng et al. 2017). Likewise, six genomic regions could be associated with salinity tolerance through analyzing soybean with SoySNP50K iSelect BeadChip (Huang et al. 2018). A more recent genotyping-by-sequencing (GBS) analysis in cowpea led authors to discover association of SNP markers with salinity tolerance at germination and seedling stages (Ravelombola et al. 2017). Large-scale and accurate phenotyping protocols may further increase the efficiency of GWAS and, the stable and consistent SMTAs could be deployed in genomics-assisted breeding for improving salinity tolerance in various grain legumes.
Genome sequencing/re-sequencing to reveal novel functional variants
Latest developments in genome sequencing/ re-sequencing motivated by next generation sequencing (NGS) chemistry have opened up promising avenues to uncover functional genetic diversity associated with traits of agricultural significance (Bohra and Singh 2015; Jha et al. 2016). Sequencing whole genomes holds great importance for capturing the genetic diversity harboured particularly in the wild accessions or landraces, which might have lost during domestication (Qi et al. 2014; Patil et al. 2016). Noteworthy progress has been made to elucidate novel genomic variants associated with the traits of breeding interest in different grain legumes through genome sequencing (Lam et al. 2010; Li et al. 2013, 2014; Varshney et al. 2013; Qi et al. 2014; Schmutz et al. 2014; Zhou et al. 2015; Patil et al. 2016; Rendón-Anaya et al. 2017). A recent sequencing attempt of 31 landraces and 22 wild soybeans elucidated nine haplotypes containing two salt-tolerant and seven salt-sensitive (Guan et al. 2014a). The authors also confirmed the widest geographical distribution of salt tolerance causative H1 haplotype in soybean. Furthermore, re-sequencing of more than 100 soybean lines enabled identification of three major structural variants namely SV1, SV2 and SV3 in the promoter and coding sequences of GmCHX1 gene (Patil et al. 2016). Importantly, the SV1 manifested by the salt-sensitive genotypes W 82 and C 08 contained Ty1/copia retrotransposon in the given locus (Qi et al. 2014), while salt tolerant SV-2 lacked Ty1/copia retrotransposon. Interestingly, the structural variant SV-3 having no retrotransposon showed salt sensitive reaction. In M. trancatula, whole genome re-sequencing of 39 wild accessions provided novel insight into the genetic basis of adaptation under salinity stress (Friesen et al. 2014). Participation of candidate gene(s) including Medtr3g098090.1 (orthologous to AtCIPK21) in abscisic acid and jasmonic acid signal transduction pathway was shown (Friesen et al. 2014). Availability of sequenced reference genomes in various grain legumes including chickpea (Jain et al. 2013; Varshney et al. 2013), soybean (Schmutz et al. 2010), common bean (Schmutz et al. 2014), cowpea (Munoz-Amatriain et al. 2017) and current progress in sequencing pea genome will delineate hitherto unknown salt responsive structural variants and genes.
Availability of reference genome sequences along with re-sequencing multiple genomes of diverse lines including CWRs and landraces could help delineate novel genomic segments associated with salinity tolerance. This will eventually offer more suitable targets for genome editing techniques such as CRISPR/Cas9 (Pennisi 2013).
Diverse omics platforms to obtain causative loci
Improvements in understanding other “omes” beyond genome have helped assigning functional role to candidate gene(s)/QTL(s) that relate to complex abiotic stress including salinity stress in crop plants (Salt et al. 2008; Baxter 2009; Liu et al. 2014; Kim et al. 2015; Ahmed et al. 2016b; Conesa et al. 2016; Rathi et al. 2016).
Transcriptomic approaches to discover candidate gene(s) for salinity tolerance
Recent shift from microarrays to high-throughput RNA sequencing (RNA-seq) technology has accelerated global expression profiling of the candidate gene(s) that respond to stress (Liu et al. 2014; Vu et al. 2015; Conesa et al. 2016). These technologies enhance possibilities to decipher transcripts/genes that play pivotal role in “regulation of transcriptional and translational machineries” under salt stress (Sahi et al. 2006a, b; Jamil et al. 2011).
An array of functional genomic resources have been established for analyzing the expression patterns of various salinity tolerance genes in chickpea; such as cDNA- microarray (Mantri et al. 2007), expressed sequenced tags (ESTs) (Varshney et al. 2009) and deep super serial analysis of gene expression (SAGE) (Molina et al. 2011). Subsequently, RNA-seq analysis of root tissue at vegetative and reproductive stages under salt stress revealed 1376 and 3660 DEGs, respectively in chickpea. Concerning gene function, majority of these differentially expressed genes (DEGs) were associated with cellular metabolic processes (carbohydrate and lipid metabolism), and cell redox homeostasis to ethylene hormone signaling under salinity stress (Garg et al. 2016). Expression analysis of GmPAP3 gene induced under salt stress suggested its possible contribution to salt tolerance in soybean through reactive oxygen species (ROS) scavenging (Liao et al. 2003). Higher induction of cellular antioxidant genes and differential expression of alternative oxidase 1(Aox1) gene in root of M. trancatula could play significant role in salt stress adaptation (Mhadhbi et al. 2011, 2013). Differential expressions of TF genes such as bHLH-type, ZFP, bZIP, YABBY, HD-Zip, ERF/AP2, WRKY are notable owing to their role in regulating plant response to abiotic stresses including salinity stress (de Lorenzo et al. 2007; Merchan et al. 2007; Sanchez et al. 2009; Song et al. 2012; Zahaf et al. 2012; Chen et al. 2014; Deinlein et al. 2014; Zhao et al. 2017). Notably, over expression of MtCBF4 in M. truncatula (Li et al. 2011) and higher expression of Zpt2-2 and CBF4 TFs in three annual Medicago genotypes (Mokhtari et al. 2017) rendered them salinity tolerant. In common bean, Hiz et al. (2014) obtained differential expression of 6422 and 4555 unigenes from leaf and root tissues, respectively along with 441 salt responsive TFs from 2678 putative TFs under salinity stress (Table 4). A total of 155 bHLH genes were recovered via comprehensive genome wide analysis in common bean (Kavas et al. 2016). Importantly, expression analysis confirmed differential expression of 63 PvbHLH genes under salinity stress. Additionally, the authors employed RNA-seq analysis to illustrate the participatory role of miRNA belonging to PvHLH-22 and PvHLH-44 genes under salt stress. Gene expression profiling under salt stress revealed up regulation of 65 GmWRKY genes and down regulation of WRKY71 gene in soybean (Yu et al. 2016). While, 47 GmWRKY and two WRKY genes showed down and up regulation, respectively in RNA-seq analysis of aerial part of plant under salt stress in soybean. Similarly in chickpea, differential expression was observed for WRKY-TF genes under salinity stress (Garg et al. 2016). More recently, function of miR156 in response to salinity stress was demonstrated in alfalfa (Arshad et al. 2017). Authors proposed that miR156 led suppression of SPL TFs in miR156-overexpressing alfalfa possibly serves as an activator for expression of non-SPL TFs. This in turn causes over-expression of various downstream genes including NHX1, HK1 HC-ATPase and SOS1, which eventually improves salinity tolerance of alfalfa via increased biomass, reduced Na+ toxicity, etc. With growing refinements in sequencing technologies, contribution of non-coding RNA (nc RNA) molecules including both long non coding RNA (lnc RNA) and small RNA (sRNA) or micro RNA (miRNA) in acclimatization of plants to a range of abiotic stresses is evident (Long et al. 2015; Liu et al. 2015). A plethora of salt responsive sRNAs regulating plant gene expression programme in response to salinity stress have been disclosed (Liu et al. 2015; Long et al. 2015). Recently, Pan et al. (2016) unveiled six target genes namely Glyma.10G116600, Glyma.02G087400, Glyma.13G329700, Glyma.12G073300, Glyma.15G044400, and Glyma.11G053800 for miR172a expressed under salt stress in soybean (Table 4). It was also postulated the underlying mechanistic role of miR172a in salt tolerance via cleaving AP2/EREBP-type TF gene SSAC1, triggering induction of thiamine biosynthesis gene THI1 encoding positive regulator for salt tolerance. Majority of the regulatory genes are involved in basic biological metabolic processes covering photosynthesis, biosynthesis of unsaturated fatty acids to phenylalanine metabolism. By employing a comparative genomics approach in cowpea, 18 conserved miRNAs and corresponding 15 target genes were recovered under salt stress (Paul et al. 2011). Of these 15 genes, upregulation of seven genes under salt stress was validated through qRT-PCR assay. While, Kohli et al. (2014) recovered miRNAs miR156_1 and miR156_10 showing low up regulation and, novel miRNAs viz., car-miR008, car-miR011 and car-miR015 showing three fold up regulation under salt stress in chickpea using high throughput sequencing of ICC 4958 RNA. The target gene of miR156 was reported to encode squamosa promoter-binding protein. To quantify and localize gene expression simultaneously, Lieben et al. (2017) suggested ‘spatial transcriptomic’ approach that combines data from histological imaging and RNA sequencing. The global transcriptome profiling improves our capacity to understand the gene expression networks, regulatory molecules including TFs and the crosstalk between different pathways that participate to impart salinity tolerance in plants.
Proteomics
Proteomics allows comprehending the changes in proteins participating in essential biological pathways and post translational modifications of ‘stress-induced’ proteins, essentially needed by plants to acclimatize themselves to various abiotic stresses (Kosová et al. 2011; Hossain et al 2013; Ramalingam et al. 2015). Thus, it could be of paramount significance to capture the entire proteins produced in response to various abiotic stresses including salt stress, thus furthering our knowledge about the protein networks associated with salt responsive signaling pathways (Ji et al. 2016). Importantly, various strategies deployed by plants to adapt themselves under salt stress at cellular, metabolic and whole plant level could be deciphered via proteomics (Ahmad et al. 2016; Silveira and Carvalho 2016; Zargar et al. 2017). Among legume crops, comprehensive proteomic studies are mostly available in two major legume crops viz., soybean and M.truncatula (Rathi et al. 2016). Following salinity stress changes occur in crucial proteins including those related to photosynthesis, carbohydrate and energy metabolism, anti oxidant/ROS scavenging, and signal transduction pathways (Long et al. 2016) (for details see Kumari et al. 2015). Concerning changes in proteins in photosynthesis, differential expression was noted in soybean leaves, hypocotyls, and roots in response to salt stress (Sobhanian et al. 2010). Down regulated proteins included ‘glyceraldehyde-3-phosphate dehydrogenase’ (expressed in leaf/hypocotyls), ‘fructokinase 2’ (expressed in hypocotyls/root) that participate in photosynthesis and other metabolic processes under salinity stress (Sobhanian et al. 2010). In case of cowpea, a total of 22 differentially regulated proteins under salt stress were recovered from two contrasting genotypes (de Abreu et al. 2014). Important proteins such as ‘rubisco activase, ribulose-5-phosphate kinase (Ru5PK), oxygen-evolving enhancer (OEE) protein 2’ that participate in vital processes like energy metabolism and photosynthesis showed abundance in salt tolerant genotypes, contrary to which these key processes are negatively affected in salt sensitive cultivar limiting their growth (de Abreu et al. 2014).
MALDI-TOF/TOF mass spectrometry aimed to identify proteins pertaining to changes in cellular metabolism in salt-stressed soybean unearthed a set of 43 responsive proteins. A total of 29 proteins showed up-regulation, while 8 proteins showed down regulation and 6 proteins were recorded to be novel under salt stress (Alam et al. 2011). Importantly, understanding the role of phosphoproteins in connection to salt tolerance, proteomic analysis at various time intervals identified 2692 phosphoproteins and 5509 phosphorylation sites in contrasting soybean cultivars under salt stress (Pi et al. 2016). Additionally, phosphorylation of various TFs including MYB/MYB TF like proteins regulate the salt responsive gene(s), involved in chalcone metabolism (chalcone synthase) causing salt tolerance in soybean were recovered. Moreover, 278 and 440 salt responsive differentially expressed proteins from leave and root, respectively, were identified via iTRAQ assay in soybean under salt stress (Ji et al. 2016). Concerning their functional role, all the recorded proteins could be assigned to 13 categories covering carbohydrate metabolism, stress and defense, signaling, membrane transport to cell division. Additionally, up regulation of late embryogenesis-abundant protein, beta-conglycinin, elicitor peptide three precursor, and basic/helix-loop-helix protein, while down-regulation of protease inhibitor and lectin proteins was observed in hypocotyls and root of soybean (Aghaie et al. 2009).
Integrating proteomic data with genomic or transcriptomic data could illustrate underlying candidate gene(s) and the complex regulatory mechanisms and key signaling players involved in salinity response and tolerance in grain legumes (Ngara and Ndimba 2014).
Metabolomics
Transcriptomics and proteomics based approaches are not adequate to illuminate the global landscape of cellular response shown by plants under abiotic stress, as plant response might involve a variety of metabolites produced under stress (Hall et al. 2002; Sumner et al. 2003; Bhalla et al. 2005; Shulaev et al. 2008; Kumar et al. 2017a, b). Therefore, metabolomics is receiving increasing attention to adequately explain the “ultimate phenotype” emanating from “complex gene network and their interaction with environment” (Hill et al. 2013; Dias et al. 2015). In grain legumes, implementation of metabolomics approach, however, has remained confined to model legumes to comprehend plant stress response (Farag et al. 2008; Sanchez et al. 2008, 2010; Komatsu et al. 2011; Ramalingam et al. 2015). Like proteins, significant changes occur in various metabolites involved in carbohydrate metabolism, amino acids biosynthesis in TCA and glycolysis cycle and at other protective antioxidation system levels (Kumari et al. 2015; Jiao et al. 2018).
A comparative analysis of wild soybean W05 and cultivated soybean C08 at metabolic level suggested abundance of several compounds such as disaccharides, sugar alcohols, and acetylated amino acids in wild type than the cultivated types (Lu et al. 2013). Likewise, Zhang et al. (2016) suggested a relation between elevated levels of metabolites (phenylalanine, asparagine, citraconic acid and other metabolites derived from TCA cycle and glycolysis process) with salinity tolerance of wild type soybean (G. soja) than the cultivated type (G. max). Additionally, enhanced production of several metabolites ranging from amino acids like isoleucine, serine, l-allothreonine, glutamic acid, phenylalanine, asparagines, aspartic acid, and other fatty/organic acids and anti oxidants provides higher tolerance to salinity stress in wild soybean seedlings in comparison to cultivated soybean (Li et al. 2017b). A comparative metabolomic study in W1 common wild type soybean and W2 salinity tolerant wild type soybean revealed enhanced accumulation of different organic acids, TCA cycle metabolites and various amino acids in the root of W2 type, which in turn conferred higher salinity tolerance to W2 than W1 (Jiao et al 2018). Thus, specific metabolites produced under salinity stress could act as biomarker for distinguishing salt tolerant and sensitive genotype (Sanchez et al. 2011). However, reports on elucidating the role of various metabolites involved in salinity acclimation are limited in grain legumes.
Ionomics
Ionomics is one of the emerging ‘omics’ approaches that facilitates ‘high throughput elemental profiling’of an organism (Salt et al. 2008; Baxter 2009). Ionomics could decipher the role of mineral nutrients dynamics causing plant toxicity/deficiency and thus, negatively affecting different plant growth stages under various abiotic stresses including salinity (Shelden and Roessner 2013). Also, ionomics can be employed as both forward and reverse genetic tool for investigating gene(s) function in plant (Baxter 2009). A number of high throughput techniques such as inductively coupled plasma-mass spectrometry (ICP-MS), inductively coupled plasma-atom/optical emission spectrometry (ICP-AES/OES), X-ray fluorescence (XRF), laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), neutron activation analysis (NAA) have been used for studying ionomes (Becker 2010; Sanchez et al. 2011; Wu et al. 2013). Ionomics has also been applied to elucidate ion detoxification and ion homeostasis in response to salinity in cereals (Wu et al. 2013) and legumes such as Lotus creticus, an extremophile and L.corniculatus and L.tenuis, glycophytic legume species (Sanchez et al. 2011). The study showed differential expression of Ca, Mg, Mn, Fe, and Zn in plants in response to salinity stress. In parallel, considerable elemental changes such as an increase in S, P and Zn was shown by salinity tolerant genotypes in comparison to salinity sensitive genotypes. Changes in Na+, Zn+2, Ca+2, and Mg+2 ions in miR156-overexpressing alfalfa genotypes at different levels of salinity stress led authors to propose that overexpressive miR156 alfalfa accumulate lower Na+ to manifest salinity tolerance (Arshad et al. 2017). In recent years, association between altered content of various elements and salinity response has been demonstrated in various crops. For example, in maize dynamics of various essential nutrient elements ranging from K, Na, Ca, Zn, Fe, Cu, Mn to B in root and shoot seedling under salinity was reported (Guo et al. 2017). Importantly, abundant Na+ and Cl− content in both old and young leaves and a decrease in K content in older leaves were noticed. The content of Ca was highest in old leaves, while K content was highest in the young leaves under alkali stress in cotton (Guo et al. 2016). The immense potential of ionomics techniques in deciphering salinity stress tolerance remains to be seen in case of grain legumes.
Phenomics to describe physiological response to salinity stress
Despite unprecedented progress in genomic data generation, phenotyping capacity still offers a key bottleneck to understand the quantitatively inherited complex traits that are highly influenced by the environment (Furbank and Tester 2011). Enhanced precision and throughput of trait phenotyping including response to salinity, high- throughput phenotyping platforms have been recently established; examples include The Plant Accelerator in Adelaide, Australia and International Plant Phenotyping Network (http://www.plant-phenotyping.org/) and so forth. These next-generation phenotyping techniques offer several advantages over conventional phenotype recording such as non-destructive measurement/ non-invasive phenotyping, automation of data, spectral imaging of complex traits, generating precise records such as ‘ionic responses’ under salinity stress (Berger et al. 2012; Campbell et al. 2015; Al-Tamimi et al. 2016; Negrao et al. 2017). Image-based phenotyping approach including thermal infrared imaging, fluorescence imaging, 3D imaging (James and Sirault 2012; Li et al. 2014; Campbell et al. 2015; Fahlgren et al. 2015; Atieno et al. 2017) is emerging as one of the important high throughput phenotyping platforms to record the minute changes in morphological and physiological responses at ‘high time and spatial resolution’ (Negrao et al. 2017). These responses range from ‘chlorophyll fluorescence and leaf water content’, relative growth rate to leaf senescence under salinity stress in large set of germplasm or in mapping population at a time (Berger et al. 2012; Hairmensis et al. 2014; Campbell et al. 2015; Awlia et al. 2016). In this regard, relative growth rate dynamics measured at various time scales using high-resolution imaging system (fixed 5 megapixel visible/RGB camera) under the salinity stress in chickpea is noteworthy (Atieno et al. 2017). As discussed by Negrao et al. (2017), high throughput phenotyping in plants enables accurate assessment of effects of salt stress at various physiological levels including photosynthesis, transpiration, ionic relation, plant senescence and also on yield and other traits relevant to salinity tolerance. Advances in plant phenomics including automated and digital imaging may further increase our understanding of the diverse temporal response of genotype under salinity stress. In this regard, ‘deep learning’ (Singh et al. 2018), ‘active vision cell’ (AVC) image-acquisition (Gibbs et al. 2018) and other modern phenotyping tools could be employed for identifying, quantifying and predicting plant salinity response with enhanced precision.
Marker-assisted selection (MAS) for salinity tolerance: some examples in grain legumes
Trait mapping studies in grain legumes have led to development of breeder-friendly genotyping assays to allow QTL introgression into elite cultivars through simple MAS or marker assisted back crossing (MABC) technique (Collard and Mackill 2008). With regard to MAS for salinity tolerance, noteworthy examples are now available across different crops (for details see Asraf and Foolad 2013; Arzani and Ashraf 2016). MABC enabled transfer of Ncl gene from wild soybean JWS 156-1 to salt sensitive cultivar Jackson underscores the immense potential of DNA marker technology for targetted and accelerated improvement of salt stress tolerance in grain legumes (Do et al. 2016). The resulting introgression lines with the Ncl gene introgressed in them showed up to 5.5 times higher yield under salinity stress. Greater efficiency of MAS for enhancing salt tolerance was demonstrated in soybean breeding programs with DNA markers Barcsoyssr_3_1306, Barcsoyssr_3_1310 and QS080465 InDel registering 76.2%, 90.1% and 94.3% efficiencies, respectively (Guan et al. 2014b). Similarly, Liu et al. (2016) successfully employed MAS for differentiating between salt-tolerant and salt-sensitive ILs (Tiefeng 8 × 85–140) using an InDel marker. Though cases of MAS for improved salinity tolerance have been reported only in soybean, growing number of MTAs reported in different grain legumes hold promise to extend usage of these technologies to other less researched crops (Do et al. 2016; Liu et al. 2016).
Emerging role of epigenetics in understanding salinity stress
Recent technological advancement has greatly supported epigenetic regulation as “mechanistic basis of stress memory” that plays a greater role in regulating various stress responsive gene(s) for plant survival under various environmental stresses including salinity stress (Kim et al. 2010, 2015; Pandey et al. 2016a, b; Lamke and Baurle 2017). Role of epigenetics in conferring ‘phenotypic plasticity’ to plants to adapt themselves to various abiotic stresses has been critically reviewed (Gallusci et al. 2017). Regulation of various important TF coding genes via epigenetic modification (methylation and histone modification) could play significant role in adapting soybean under salinity stress (Song et al. 2012). Out of 10 TFs, significant up regulatory induction in four TFs (one MYB, one b-ZIP and two AP2/DREB) gene such as Glyma11g02400, Glyma08g41450, Glyma16g27950 and Glyma20g30840 were noted due to changes in methylation pattern in the promoter regions of the given genes under salinity stress (Song et al. 2012). Methylation and demethylation status of H3K4me3, H3K9me2 and H3K9ac control the expression of these genes under salinity stress (Song et al. 2012). Differentially methylated regions and their relevant role in expression of various genes related to abiotic stresses including salinity stress has been recently uncovered via whole genome bisulphate sequencing in rice (Garg et al. 2015; Wang et al. 2015), wheat (Wang et al. 2014; Kumar et al. 2017a, b), brassica (Marconi et al. 2013), maize (Forestan et al. 2016). More research on epigenetics will increasingly uncover the association between ‘epigenetic biomarker’ and alterations in gene expressions in response to salinity stress. However, progress of understanding of salinity stress response in grain legume at epigenetic level remains limited.
Integration of “omics” to sustain grain legume yield under salinity stress
With the deluge of high-throughput‘omics’ data, bioinformatics/computational biology is receiving greater attention to derive better inferences from the large-scale datasets (Colmsee et al. 2012; Deshmukh et al. 2014; Jha et al. 2017). However, integration of these vast ‘omics’ data to resolve the complex traits at functional level poses a big challenge (Deshmukh et al. 2014). A complex network of signal transduction pathways and several levels of regulations occurring at gene regulation, transcription, post-transcription and post-translational render salinity a complex trait (Li et al. 2017a, b). Thus, ‘omics’ including transcriptomics, proteomics, metabolomics, ionomics, and epigenomics level ultimately, decides the phenotypic expression of gene(s) contributing in salinity stress tolerance. To retrieve information pertaining to gene, genomics, transcriptomics, proteomics and other ‘omics’, several web-based resources have been established in various legume such as Chickpea Transcriptome Database (CTDB) in chickpea (Verma et al. 2015), Cowpea Genespace/Genomics Knowledge Base (CGKB) in cowpea (Chen et al. 2007), Soybean Knowledge Base in soybean (SKB, http://soykb.org), and Legume information system (LegumeInfo. org) (Dash et al. 2016). To expeditiously breed tolerant cultivars in grain legumes, deployment of candidate gene(s)/QTL(s) contributing salinity tolerance through effective integration of multilayer ‘omics’ science and breeding techniques is illustrated in Fig. 1.

Integrated ‘omics’ approaches for development of salinity tolerant genotypes
Conclusion and future directions
Yield gains of grain legume crops need incremental increase in the face of growing food demands coupled with an expansion in salt affected area (Alexandratos and Bruinsma 2012). Large-scale deployment of CWRs is imperative to bring ‘salinity adaptive traits’ into the cultivated pool. This, however, will require: (a) reliable evaluation of germplasm resources for salinity tolerance (b) monitoring precise phenotypic response of genotypes at various growth stages under salinity stress (Atieno et al. 2017) and (c) multi-location field testing of tolerant genotypes under salinity stress. To support crop improvement programs, ‘multi-layered omics science’ could play a significant role in deciphering plant salinity response. In addition, new breeding techniques such as ‘speed breeding’ (Li et al. 2018), genomic selection (Goddard and Hayes 2007) and genome editing via CRISPR-Cas9 (Pennisi 2013) could enable faster development of stress tolerant plants. The improved knowledge about plant salinity response coupled with more efficient breeding programs will certainly contribute to sustain productivity levels of grain legume crops under salt-stressed scenario.
Author contribution statement
UCJ conceived the idea and wrote the MS with AB. AB and SKP corrected the language of the MS. RJ conducted literature survey. All authors have read and approved the final manuscript.
References
Abel GH (1969) Inheritance of the capacity for chloride inclusion and chloride exclusion by soybeans. Crop Sci 9:697–698
Acosta-Motos JR, Ortuño MF, Bernal-Vicente A, Diaz-Vivancos P, Sanchez-Blanco MJ, Hernandez JA (2017) Plant responses to salt stress: adaptive Mechanisms. Agron 7:18
Aghaei K, Ehsanpour AA, Shah AH, Komatsu S (2009) Proteome analysis of soybean hypocotyl and root under salt stress. Amino Acids 36:91–98
Ahmad P, Jhon R (2005) Effect of salt stress on growth and biochemical parameters of Pisum sativum L. Arch Agron Soil Sci 51:665–672
Ahmad P, Abdel Latef AA, Rasool S, Akram NA, Ashraf M, Gucel S (2016) Role of proteomics in crop stress tolerance. Front Plant Sci 7:1336
Alam I, Sharmin SA, Kim KH, Kim YG, Lee JJ, Bahk JD, Lee BH (2011) Comparative proteomic approach to identify proteins involved in flooding combined with salinity stress in soybean. Plant Soil 346:45
Alexandratos N, Bruinsma J (2012) World agriculture towards 2030/2050: The 2012 revision. ESA Working paper No. 12-03. Food and Ag. Org. United Nations, Rome
Al-Mutawa MM (2003) Effect of salinity on germination and seedling growth of chickpea (Cicer arietinum L.) genotypes. Int J Agric Biol 5:226–229
Al-Tamimi N, Brien C, Oakey H, Berger B, Saade S, Ho YS, Schmöckel SM, Tester M, Negrão S (2016) Salinity tolerance loci revealed in rice using high-throughput non-invasive phenotyping. Nat Commun 7:1–11
Amitai-Zeigersona H, Scolnikb PA, Bar-Zvi D (1995) Tomato Asrl mRNA and protein are transiently expressed following salt stress, osmotic stress and treatment with abscisic acid. Plant Sci 110:205–213
Arshad M, Gruber MY, Wall K, Hannoufa A (2017) An insight into microRNA156 role in salinity stress responses of alfalfa. Front Plant Sci 8:356
Arzani A, Ashraf M (2016) Smart engineering of genetic resources for enhanced salinity tolerance in crop plants. Crit Rev Plant Sci 35:146–189
Ashraf M (2004) Some important physiological selection criteria for salt tolerance in plants. Flora 199:361–376
Ashraf M, Foolad MR (2013) Crop breeding for salt tolerance in the era of molecular markers and marker-assisted selection. Plant Breed 132:10–20
Ashraf M, Harris PJC (2004) Potential biochemical indicators of salinity tolerance in plants. Plant Sci 166:3–16
Ashraf M, Waheed A (1993) Responses of some genetically diverse lines of chickpea (Cicer arietinum L.) to salt. Plant Soil 154:257–266
Ashraf M, Waheed A (1998) Components of genetic variation of salt tolerance in chickpea (Cicer arietinum L.). Arch Agron Soil Sci 42:415–424
Ashraf M, Wu L (2011) Breeding for salinity tolerance in plants. Crit Rev Plant Sci 13:17–42
Atieno J, Li Y, Langridge P, Dowling K, Brien C, Berger B, Varshney RK, Sutton T (2017) Exploring genetic variation for salinity tolerance in chickpea using image-based phenotyping. Sci Report 7:1300
Awlia M, Nigro A, Fajkus J, Schmoeckel SM, Negrão S, Santelia D, Trtílek M, Tester M, Julkowska MM, Panzarová K (2016) High-throughput non-destructive phenotyping of traits that contribute to salinity tolerance in Arabidopsis thaliana. Front Plant Sci 7:1414
Banzai T, Hershkovits G, Katcoff DJ, Hanagata N, Dubinsky Z, Karube I (2002) Identification and characterization of mRNA transcripts differentially expressed in response to high salinity by means of differential display in the mangrove, Bruguiera gymnorrhiza. Plant Sci 162:499–505
Baxter I (2009) Ionomics: studying the social network of mineral nutrients. Curr Opin Plant Biol 12:381–386
Bayuelo-Jiménes JS, Craig R, Lynch JP (2002a) Salinity tolerance of Phaseolus species during germination and early seedling growth. Crop Sci 42:1584–1594
Bayuelo-Jiménes JS, Debouck DG, Lynch JP (2002b) Salinity tolerance of Phaseolus species during early vegetative growth. Crop Sci 42:2184–2192
Bayuelo-Jiménes JS, Debouck DG, Lynch JP (2003) Growth, gas exchange, water relations, and ion composition of Phaseolus species grown under saline conditions. Field Crops Res 80:207–222
Becker JS (2010) Imaging of metals, metalloids, and non-metals by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) in biological tissues. In: Rubakhin SS, Sweedler JV (eds) Mass spectrometry imaging: principles and protocols. Humana Press, Totowa, pp 51–82
Berger B, de Regt B, Tester M (2012) Trait dissection of salinity tolerance with plant phenomics. Methods Mol Biol 913:399–413
Bhalla R, Narasimhan K, Swarup S (2005) Metabolomics and its role in understanding cellular responses in plants. Plant Cell Rep 24:562–571
Bohra A (2013) Emerging paradigms in genomics-based crop improvement. Sci World J 585467:17
Bohra A, Singh NP (2015) Whole genome sequences in pulse crops: a global community resource to expedite translational genomics and knowledge-based crop improvement. Biotechnol Lett 37:1529–1539
Bohra A, Pandey MK, Jha UC, Singh B, Singh IP, Datta D, Chaturvedi SK, Nadarajan N, Varshney RK (2014) Genomics assisted breeding in four major pulse crops of developing countries: present status and prospects. Theor Appl Genet 127:1263–1291
Bohra A, Sahrawat KL, Kumar S, Joshi R, Parihar AK, Singh U, Singh D, Singh NP (2015) Genetics- and genomics-based interventions for nutritional enhancement of grain legume crops: status and outlook. J Appl Genet 56:151–161
Broughton WJ, Hernandez G, Blair M, Beebe S, Gepts P, Vanderleyden J (2003) Beans (Phaseolus spp.): model food legumes. Plant Soil 252:55–128
Brozynska M, Furtado A, Henry RJ (2016) Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol J 14:1070–1085
Büyük İ, Inal B, Ilhan E, Tanriseven M, Aras S, Erayman M (2016) Genome-wide identification of salinity responsive HSP70s in common bean. Mol Biol Rep 43:1251–1266
Cabot C, Sibole JV, Barceló J, Poschenrieder C (2014) Lessons from crop plants struggling with salinity. Plant Sci 226:2–13
Campbell MT, Knecht AC, Berger B, Brien CJ, Wang D, Walia H (2015) Integrating image-based phenomics and association analysis to dissect the genetic architecture of temporal salinity responses in rice. Plant Physiol 168:1476–1697
Chankaew S, Isemura T, Naito K, Ogiso-Tanaka E, Tomooka N, Somta P, Kaga A, Vaughan DA, Srinives P (2014) QTL mapping for salt tolerance and domestication-related traits in Vigna marina subsp. oblonga, a halophytic species. Theor Appl Genet 127:691–702
Chen TH, Murata N (2011) Glycinebetaine protects plants against abiotic stress: mechanisms and biotechnological applications. Plant Cell Environ 34:1–20
Chen XQ, Yu BJ (2007) Ionic effects of Na+ and Cl– on photosynthesis in Glycine max seedlings under isoosmotic salt stress. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao 33:294–300
Chen X, Laudeman TW, Rushton PJ, Spraggins TA, Timko MP (2007) CGKB: an annotation knowledge base for cowpea (Vigna unguiculata L.) methylation filtered genomic genespace sequences. BMC Bioinform 8:129
Chen H, Cui S, Fu S, Gai J, Yu D (2008) Identification of quantitative trait loci associated with salt tolerance during seedling growth in soybean (Glycine max L.). Austr J Agric Res 59:1086–1091
Chen JB, Wang SM, Jing RL, Mao XG (2009) Cloning the PvP5CS gene from common bean (Phaseolus vulgaris) and its expression patterns under abiotic stresses. J Plant Physiol 166:12–19
Chen P, Yan K, Shao H, Zhao S (2013) Physiological mechanisms for high salt tolerance in wild soybean (Glycine soja) from Yellow River Delta, China: photosynthesis, osmotic regulation, ion flux and antioxidant capacity. PLoS One 8:e83227
Chen X, Chen Z, Zhao H, Zhao Y, Cheng B, Xiang Y (2014) Genome-wide analysis of soybean HD-Zip gene family and expression profiling under Salinity and drought treatments. PLoS One 9:e87156
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci 363:557–572
Colmsee C, Mascher M, Czauderna T, Hartmann A, Schlüter U, Zellerhoff N, Schmitz J, Bräutigam A, Pick TR, Alter P, Gahrtz M, Witt S, Fernie AR, Börnke F, Fahnenstich H, Bucher M, Dresselhaus T, Weber AP, Schreiber F, Scholz U, Sonnewald U (2012) OPTIMAS-DW: a comprehensive transcriptomics, metabolomics, ionomics, proteomics and phenomics data resource for maize. BMC Plant Biol 12:245
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A (2016) A survey of best practices for RNA-seq data analysis. Genome Biol 17:13
Considine MJ, Siddique KHM, Foyer CH (2017) Nature’s pulse power: legumes, food security and climate change. J Expt Bot 68:1815–1818
Costa JH, Jolivet Y, Hasenfratz-Sauder MP, Orellano EG, da Guia Silva Lima M, Dizengremel P, Fernandes de Melo D (2007) Alternative oxidase regulation in roots of Vigna unguiculata cultivars differing in drought/salt tolerance. J Plant Physiol 164:718–727
Dash S, Campbell JD, Cannon EK, Cleary AM, Huang W, Kalberer SR, Karingula V, Rice AG, Singh J, Umale PE, Weeks NT, Wilkey AP, Farmer AD, Cannon SB (2016) Legume information system (LegumeInfo. org): a key component of a set of federated data resources for the legume family. Nucl Acids Res 44:D1181–D1188
de Lorenzo F, Merchan S, Blanchet M, Megiàs F, Frugier M, Crespi CS (2007) Differential expression of the TFIIIA regulatory pathway in response to salt stress between Medicago truncatula genotypes. Plant Physiol 145:1521–1532
de Abreu CE, Araújo Gdos S, Monteiro-Moreira AC, Costa JH, Leite Hde B, Moreno FB, Prisco JT, Gomes-Filho E (2014) Proteomic analysis of salt stress and recovery in leaves of Vigna unguiculata cultivars differing in salt tolerance. Plant Cell Rep 33:1289–1306
Deinlein U, Stephan AB, Horie T, Luo W, Xu G, Schroeder JI (2014) Plant salt-tolerance mechanisms. Trend Plant Sci 19:371–379
DeRose-Wilson L, Gaut BS (2011) Mapping salinity tolerance during Arabidopsis thaliana germination and seedling growth. PLoS One 6:e22832
Deshmukh R, Sonah H, Patil G, Chen W, Prince S, Mutava R, Vuong T, Valliyodan B, Nguyen HT (2014) Integrating omic approaches for abiotic stress tolerance in soybean. Front Plant Sci 5:244
Dias DA, Hill CB, Jayasinghe NS, Atieno J, Sutton T, Roessner U (2015) Quantitative profiling of polar primary metabolites of two chickpea cultivars with contrasting responses to salinity. J Chromatogr B Anal Technol Biomed Life Sci 1000:1–13
Do TD, Chen H, Hien VT, Hamwieh A, Yamada T, Sato T, Yan Y, Cong H, Shono M, Suenaga K, Xu D (2016) Ncl synchronously regulates Na+, K+, and Cl– in soybean and greatly increases the grain yield in saline field conditions. Sci Rep 6:19147
Dua RP (1992) Differential response of chickpea (Cicer arietinum) genotypes to salinity. J Agric Sci 119:367–371
Dua RP (1998) Salinity tolerance in chickpea (Cicer arientinum L.). Indian J Plant Physiol 3:102–106
Dua RP, Sharma PC (1995) Salinity tolerance of Kabuli and Desi chickpea genotypes. Intl Chickpea Pigeonpea Newslett 2::19–22
Durand M, Lacan D (1994) Sodium partitioning within the shoot of soybean. Physiol Plant 91:65–71
Essa TA (2002) Effect of salinity stress on growth and nutrient composition of three soybean (Glycine max L. Merrill) cultivars. J Crop Agron Crop Sci 188:86–93
Fahlgren N, Gehan MA, Baxter I (2015) Lights, camera, action: high-throughput plant phenotyping is ready for a close-up. Curr Opin Plant Biol 24:93–99
FAO (2015) FAO land and plant nutrition management service. Food and Agriculture Organization of the United Nations, Rome
Farag MA, Huhman DV, Dixon RA, Sumner LW (2008) Metabolomics reveals novel pathways and differential mechanistic and elicitor-specific responses in phenylpropanoid and isoflavonoid biosynthesis in Medicago truncatula cell cultures. Plant Physiol 146:387–402
Farooq M, Gogoi N, Hussain M, Barthakur S, Paul S, Bharadwaj N, Migdadi HM, Alghamdi SS, Siddique KHM (2017) Effects, tolerance mechanisms and management of salt stress in grain legumes. Plant Physiol Biochem 118:199–217
Flowers TJ (2004) Improving crop salt tolerance. J Exp Bot 55:307–319
Flowers TJ, Gaur PM, Gowda CL, Krishnamurthy L, Samineni S, Siddique KH, Turner NC, Vadez V, Varshney RK, Colmer TD (2010) Salt sensitivity in chickpea. Plant Cell Environ 33:490–509
Forestan C, Cigliano RA, Farinati S, Lunardon A, Sanseverino W, Varotto S (2016) Stress-induced and epigenetic-mediated maize transcriptome regulation study by means of transcriptome reannotation and differential expression analysis. Sci Rep 6:30446
Foyer CH, Lam HM, Nguyen HT, Siddique KH, Varshney RK, Colmer TD, Cowling W, Bramley H, Mori TA, Hodgson JM, Cooper JW, Miller AJ, Kunert K, Vorster J, Cullis C, Ozga JA, Wahlqvist ML, Liang Y, Shou H, Shi K, Yu J, Fodor N, Kaiser BN, Wong FL, Valliyodan B, Considine MJ (2016) Neglecting legumes has compromised human health and sustainable food production. Nat Plants 2:16112
Freitas JBS, Chagas RM, Almeida IMR, Cavalcanti FR, Silveira JAG (2001) Expression of physiological traits related to salt tolerance in two contrasting cowpea cultivars. Documentos Embrapa Meio- Norte 56:115–118
Friesen ML, von Wettberg EJ, Badri M, Moriuchi KS, Barhoumi F, Chang PL, Cuellar-Ortiz S, Cordeiro MA, Vu WT, Arraouadi S, Djébali N, Zribi K, Badri Y, Porter SS, Aouani ME, Cook DR, Strauss SY, Nuzhdin SV(2014)The ecological genomic basis of salinity adaptation in Tunisian Medicago truncatula. BMC Genom 15:1160
Furbank RT, Tester M (2011) Phenomics–technologies to relieve the phenotyping bottleneck. Trends Plant Sci 16:635–644
Gallusci P, Dai Z, Génard M, Gauffretau A, Leblanc-Fournier N, Richard-Molard C, Vile D, Brunel-Muguet S (2017) Epigenetics for plant improvement: Current knowledge and modeling Avenues. Trends Plant Sci 1360–1385:30089–30090
Gama PBS, Inanaga S, Tanaka K, Nakazawa R (2007) Physiological response of common bean (Phaseolus vulgaris L.) seedlings to salinity stress. Afr J Biotechnol 6:079–088
Garg R, Narayana V, Shankar R, Jain M (2015) Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci Rep 5:14922
Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, Varshney RK, Bhatia S, Jain M (2016) Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci Rep 6:19228
Gibbs JA, Pound M, French AP, Wells DM, Murchie E, Pridmore T (2018) Plant phenotyping: an active vision cell for three-dimensional plant shoot reconstruction. Plant Physiol 178:524–534
Goddard ME, Hayes BJ (2007) Genomic selection. J Anim Breed Genet 124:323–330
Goerzt SH, Coons JM (1991) Tolerance of tepary and navy beans to NaCl during germination and emergence. Hort Sci 24:249–293
Gogile A, Andargie M, Muthuswamy M (2013) Screening selected genotypes of cowpea [Vigna unguiculata (L.) Walp.] for salt tolerance during seedling growth stage. Pak J Biol Sci 16:671–679
Greenland DJ (1984) Exploited plants: rice. Biologist 31:291–325
Guan R, Qu Y, Guo Y, Yu L, Liu Y, Jiang J, Chen J, Ren Y, Liu G, Tian L, Jin L, Liu Z, Hong H, Chang R, Gilliham M, Qiu L (2014a) Salinity tolerance in soybean is modulated by natural variation in GmSALT3. Plant J 80:937–950
Guan R, Chen J, Jiang J, Liu G, Liu Y, Tian L, Yu L, Chang R, Qiu L (2014b) Mapping and validation of a dominant salt tolerance gene in the cultivated soybean (Glycine max) variety Tiefeng8. Crop J 2:358–365
Guo R, Shi LX, Yang CW, Yan CR, Zhong XL, Liu Q, Xia X, Li HR (2016) Comparison of ionomic and metabolites response under alkali stress in old and young leaves of cotton (Gossypium hirsutum L.) seedlings. Front Plant Sci 7:1785
Guo R, Shi L, Yan C, Zhong X, Gu F, Liu Q, Xia X, Li H (2017) Ionomic and metabolic responses to neutral salt or alkaline salt stresses in maize (Zea mays L.) seedlings. BMC Plant Biol 17:41
Gupta B, Huang B (2014) Mechanism of salinity tolerance in plants: physiological, biochemical, and molecular characterization. Intl J Genom 2014:1–18
Ha BK, Vuong TD, Velusamy V, Nguyen HT, Shannon JG, Lee JD (2013) Genetic mapping of quantitative trait loci conditioning salt tolerance in wild soybean (Glycine soja) PI 483463. Euphytica 193:79–88
Hairmansis A, Berger B, Tester M, Roy SJ (2014) Image-based phenotyping for non-destructive screening of different salinity tolerance traits in rice. Rice 6:16
Hall R, Beale M, Fiehn O, Hardy N, Sumner L, Bino R (2002) Plant metabolomics: the missing link in functional genomics strategies. Plant Cell 14:1437–1440
Hamwieh A, Xu DH (2008) Conserved salt tolerance quantitative trait locus (QTL) in wild and cultivated soybeans. Breed Sci 58:355–359
Hamwieh A, Tuyen DD, Cong H, Benitez ER, Takahashi R, Xu DH (2011) Identification and validation of a major QTL for salt tolerance in soybean. Euphytica 179:451–459
Hernández JA, Almansa MS (2002) Short-term effects of salt stress on antioxidant systems and leaf water relations of pea leaves. Physiol Plant 115:251–257
Hernández JA, Corpas FJ, Gómez M, del Río LA, Sevilla F (1993) Salt induced oxidative stress mediated by activated oxygen species in pea leaf mitochondria. Physiol Plant 89:103–110
Hernández JA, Jiménez A, Mullineaux P, Sevilia F (2000) Tolerance of pea (Pisum sativum L.) to long-term salt stress is associated with induction of antioxidant defences. Plant Cell Environ 23:853–862
Hill CB, Roessner U (2013) Metabolic Profiling of Plants by GC-MS. In: Weckwerth W, Kahl G (eds) The handbook of plant metabolomics: metabolite profiling and networking, first edn. Wiley, Weinheim, pp 3–23
Hiz MC, Canher B, Niron H, Turet M (2014) Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One 9:e92598
Hossain Z, Khatoon A, Komatsu S (2013) Soybean proteomics for unraveling abiotic stress response mechanism. J Proteome Res 12:4670–4684
Hoyos-Villegas V, Song Q, Kelly JD (2017) Genome-wide association analysis for drought tolerance and associated traits in common Bean. Plant Genome https://doi.org/10.3835/plantgenome2015.12.0122
Huang L, Zeng A, Chen P, Wu C, Wang D, Wen Z (2018) Genomewide association analysis of salt tolerance in soybean [Glycine max (L.) Merr.]. Plant Breed. https://doi.org/10.1111/pbr.12623
Ismail AM, Horie T (2017) Genomics, physiology, and molecular breeding approaches for improving salt tolerance. Annu Rev Plant Biol 68:19.1–19.30
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G, Yadav M, Kant C, Sharma P, Yadav G, Bhatia S, Tyagi AK, Chattopadhyay D (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729
James RA, Sirault XR (2012) Infrared thermography in plant phenotyping for salinity tolerance. Methods Mol Biol 913:173–189
Jamil A, Riaz S, Ashraf M, Foolad MR (2011) Gene expression profiling of plants under salt stress. Crit Rev Plant Sci 30:435–458
Jha UC, Barh D, Parida S, Jha R, Singh NP (2016) Applied molecular biotechnology. In Khan MS, Khan IA, Barh D (Eds), Whole-genome resequencing Current status and future prospects in crop improvement. CRC Press, Boca Raton, pp 187–212
Jha UC, Bohra A, Jha R, Parida S (2017) Integrated ‘omics’ approaches to sustain major global grain legume productivity under heat stress. Plant Breed 136:437–459
Ji W, Cong R, Li S, Li R, Qin Z, Li Y, Zhou X, Chen S, Li J (2016) Comparative proteomic analysis of soybean leaves and roots by iTRAQ provides insights into response mechanisms to short-term salt stress. Front Plant Sci 7:573
Jia Q, Xiao ZX, Wong FL, Sun S, Liang KJ, Lam HM (2017) Genome-wide analyses of the soybean F-Box gene family in response to salt stress. Int J Mol Sci 18:818
Jiao Y, Bai Z, Xu J, Zhao M, Khan Y, Hu Y, Shi L (2018) Metabolomics and its physiological regulation process reveal the salttolerant mechanism in Glycine soja seedling roots. Plant Physiol Biochem 126:187–196
Kaashyap M, Ford R, Bohra A, Kuvalekar A, Mantri N (2017) Improving salt tolerance of chickpea using modern genomics tools and molecular breeding. Curr Genom 18:557–567
Kaashyap M, Ford R, Kudapa H, Jain M, Edwards D, Varshney R, Mantri N (2018) Differential regulation of genes involved in root morphogenesis and cell wall modification is associated with salinity tolerance in chickpea. Sci Rep 8:4855
Kan GZ, Zhang W, Yang WM, Ma DY, Zhang D, Hao DR, Hu ZB, Yu DY (2015) Association mapping of soybean seed germination under salt stress. Mol Genet Genom 290:2147–2162
Kan G, Ning L, Li Y, Hu Z, Zhang W, He X, Yu D (2016) Identification of novel loci for salt stress at the seed germination stage in soybean. Breed Sci 66:530–541
Kao WY, Tsai TT, Shih CN (2003) Photosynthetic gas exchange and chlorophyll a fluorescence of three wild soybean species in response to NaCl treatments. Photosynthetica 41:415–419
Kao WY, Tsai TT, Tsai HC, Shih CN (2006) Responses of three Glycine species to salt stress. Environ Exp Bot 56:120–125
Kavas M, Kızıldoğan AK, Gökdemir G, Baloğlu MC (2015) Genome—wide investigation and expression analysis of AP2-ERF gene family in salt tolerant common bean. Excli J 14:1187–1206
Kavas M, Baloğlu MC, Atabay ES, Ziplar UT, Daşgan HY, Ünver T (2016) Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Genet Genom 291:129–143
Khan HA, Siddique KHM, Munir R, Colmer TD (2015) Salt sensitivity in chickpea: growth, photosynthesis, seed yield components and tissue ion regulation in contrasting genotypes. J Plant Physiol 182:1–12
Khan HA, Siddique KH, Colmer TD (2016) Salt sensitivity in chickpea is determined by sodium toxicity. Planta 244:623–637
Khan HA, Siddique KHM, Colmer TD (2017) Vegetative and reproductive growth of salt-stressed chickpea are carbon-limited: sucrose infusion at the reproductive stage improves salt tolerance. J Expt Bot 68:2001–2011
Kim JM, To TK, Nishioka T, Seki M (2010) Chromatin regulation functions in plant abiotic stress responses. Plant Cell Environ 33:604–611
Kim JM, Sasaki T, Ueda M, Sako K, Seki M (2015) Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front Plant Sci 6:114
Kim MJ, Kim HJ, Pak JH, Cho HS, Choi HK, Jung HW, Lee DH, Chung YS (2017) Overexpression of AtSZF2 from Arabidopsis showed enhanced tolerance to salt stress in soybean. Plant Breed Biotech 5:1–15
Kohli D, Joshi G, Deokar AA, Bhardwaj AR, Agarwal M, Katiyar-Agarwal S, Srinivasan R, Jain PK (2014) Identification and characterization of wilt and salt stress-responsive MicroRNAs in chickpea through high-throughput sequencing. PLoS One 9:e108851
Komatsu S, Yamamoto A, Nakamura T, Nouri MZ, Nanjo Y, Nishizawa K, Furukawa K (2011) Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J Prot Res 10:3993–4004
Kosová K, Vítámvás P, Prášil IT, Renaut J (2011) Plant proteome changes under abiotic stress–contribution of proteomics studies to understanding plant stress response. J Proteom 74:1301–1322
Kotula L, Khan HA, Quealy J, Turner NC, Vadez V, Siddique KH, Clode PL, Colmer TD (2015) Salt sensitivity in chickpea (Cicer arietinum L.): ions in reproductive tissues and yield components in contrasting genotypes. Plant Cell Environ 38:1565–1577
Krishnamurthy L, Turner NC, Gaur PM, Upadhyaya HD, Varshney RK, Siddique KHM, Vadez V (2011) Consistent variation across soil types in salinity resistance of a diverse range of chickpea (Cicer arietinum L.) genotypes. J Agron Crop Sci 197:214–227
Kumar S, Beena AS, Awana M, Singh A (2017a) Salt-Induced tissue-specific cytosine methylation down regulates expression of HKT genes in contrasting wheat (Triticum aestivum L.) genotypes. DNA Cell Biol 36:283–294
Kumar R, Bohra A, Pandey AK, Pandey MK, Kumar A (2017b) Metabolomics for plant improvement: Status and prospects. Front Plant Sci 8:1302
Kumari A, Das P, Parida AK, Agarwal PK (2015) Proteomics, metabolomics, and ionomics perspectives of salinity tolerance in halophytes. Front Plant Sci 6:1–20
L’taief B, Sifi B, Zaman-Allah M, Drevon JJ, Lachaâl M (2007) Effect of salinity on root-nodule conductance to the oxygen diffusion in the Cicer arietinum-Mesorhizobium ciceri symbiosis. J Plant Physiol 164:1028–1036
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Sun SS, Zhang G (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059
Lämke J, Bäurle I (2017) Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol 18:124
Lauter DJ, Munns DN (1986) Salt resistance of chickpea genotypes in solutions salinized with NaCl or Na2SO4. Plant Soil 95:271–279
Lee GJ, Carter TE, Villagarcia MR, Li Z, Zhou X, Gibbs MO, Boerma HR (2004) A major QTL conditioning salt tolerance in S-100 soybean and descendent cultivars. Theor Appl Genet 109:1610–1619
Lee JD, Shannon JG, Vuong TD, Nguyen HT (2009) Inheritance of salt tolerance in wild soybean (Glycine soja Sieb. and Zucc.) Accession PI483463. J Hered 100:798–801
Lenis JM, Ellersieck M, Blevins DG, Sleper DA, Nguyen HT, Dunn D, Lee JD, Shannon JG (2011) Differences in ion accumulation and salt tolerance among Glycine accessions. J Agron Crop Sci 197:302–310
Leonforte A, Forster JW, Redden RJ, Nicolas ME, Salisbury PA (2013a) Sources of high tolerance to salinity in pea (Pisum sativum L.). Euphytica 189:203–216
Leonforte A, Sudheesh S, Cogan NO, Salisbury PA, Nicolas ME, Materne M, Forster JW, Kaur S (2013b) SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant Biol 13:161
Li W-Y, Wong F-L, Tsai S-N, Phang T-H, Shao G, Lam H-M (2006) Tonoplast-located GmCLC1 and GmNHX1 from soybean enhance NaCl tolerance in transgenic bright yellow (by)-2 cells. Plant Cell Environ 29:1122–1137
Li D, Zhang Y, Hu X, Shen X, Ma L, Su Z, Wang T, Dong J (2011) Transcriptional profiling of Medicago truncatula under salt stress identified a novel CBF transcription factor MtCBF4 that plays an important role in abiotic stress responses. BMC Plant Biol 11:109
Li YH, Zhao SC, Ma JX, Li D, Yan L, Li J, Qi XT, Guo XS, Zhang L, He WM, Chang RZ, Liang QS, Guo Y, Ye C, Wang XB, Tao Y, Guan RX, Wang JY, Liu YL, Jin LG, Zhang XQ, Liu ZX, Zhang LJ, Chen J, Wang KJ, Nielsen R, Li RQ, Chen PY, Li WB, Reif JC, Purugganan M, Wang J, Zhang MC, Wang J, Qiu LJ (2013) Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genom 14:579
Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, Guo Y, Zhang J, Sui Y, Zheng L, Zhang SS, Zuo Q, Shi XH, Li YF, Zhang WK, Hu Y, Kong G, Hong HL, Tan B, Song J, Liu ZX, Wang Y, Ruan H, Yeung CK, Liu J, Wang H, Zhang LJ, Guan RX, Wang KJ, Li WB, Chen SY, Chang RZ, Jiang Z, Jackson SA, Li R, Qiu LJ (2014a) De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32:1045–1052
Li L, Zhang Q, Huang D (2014b) A review of imaging techniques for plant phenotyping. Sensors 14:20078–20111
Li J, Pu L, Han M, Zhu M, Zhang R, Xiang Y (2014c) Soil salinization research in China: advances and prospects. J Geograph Sci 24:943–960
Li M, Guo R, Jiao Y, Jin X, Zhang H, Shi L (2017a) Comparison of salt tolerance in Soja based on metabolomics of seedling roots. Front Plant Sci 8:1101
Li MW, Xin D, Gao Y, Li KP, Fan K, Muñoz NB, Yung WS, Lam HM (2017b) Using genomic information to improve soybean adaptability to climate change. J Exp Bot 68:1823–1834
Li H, Rasheed A, Hickey LT, He Z (2018) Fast-forwarding genetic gain. Trends Plant Sci 23:183–186
Liang W, Ma X, Wan P, Liu L (2018) Plant salt-tolerance mechanism: a review. Biochem Biophys Res Commun 495:286–291
Liao H, Wong FL, Phang TH, Cheung MY, Li WY, Shao G, Yan X, Lam HM (2003) GmPAP3, a novel purple acid phosphatase-like gene in soybean induced by NaCl stress but not phosphorus deficiency. Gene 318:103–111
Lieben L (2017) Spatial transcriptomics in plants. Nat Rev Genet 18:394
Liu Y, Zhou J, White KP (2014) RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30:301–304
Liu X, Hao L, Li D, Zhu L, Hu S (2015) Long non-coding RNAs and their biological roles in plants. Genom Proteom Bioinform 13:137–147
Liu Y, Yu L, Qu Y, Chen J, Liu X, Hong H, Liu Z, Chang R, Gilliham M, Qiu L, Guan R (2016) GmSALT3, which confers improved soybean salt tolerance in the field, increases leaf Cl-exclusion prior to Na+ exclusion but does not improve early vigor under salinity. Front Plant Sci 7:1485
Liu A, Xiao Z, Li MW, Wong FL, Yung WS, Ku YS, Wang Q, Wang X, Xie M, Yim AK, Chan TF, Lam HM (2018) Transcriptomic reprogramming in soybean seedlings under salt stress.Plant Cell Environ. https://doi.org/10.1111/pce.13186
Long RC, Li MN, Kang JM, Zhang TJ, Sun Y, Yang QC (2015) Small RNA deep sequencing identifies novel and salt-stress-regulated microRNAs from roots of Medicago sativa and Medicago truncatula. Physiol Plant 154:13–27
Long R, Li M, Zhang T, Kang J, Sun Y, Cong L, Gao Y, Liu F, Yang Q (2016) Comparative proteomic analysis reveals differential root proteins in Medicago sativa and Medicago truncatula in response to salt Stress. Front Plant Sci 7:424
Lu KX, Cao BH, Feng XP, He Y, Jiang DA (2009) Photosynthetic response of salt-tolerant and sensitive soybean varieties. Photosyn 47:381–387
Lu Y, Lam H, Pi E, Zhan Q, Tsai S, Wang C, Kwan Y, Ngai S (2013) Comparative metabolomics in Glycine max and Glycine soja under salt stress to reveal the phenotypes of their offspring. J Agric Food Chem 36:8711–8721
Luo QY, Yu BJ, Liu YL, Zhang YM, Xue YL, Zhang Y (2004) The mixed inheritance analysis of salt tolerance in cultivars of Glycine max. Soybean Sci 23:239–244
Luo Q, Yu B, Liu Y (2005) Differential sensitivity to chloride and sodium ions in seedlings of Glycine max and G. soja under NaCl stress. J Plant Physiol 162:1003–1012
Maas EV, Poss JA (1989) Salt sensitive of cowpea at various growth stages. Irrig Sci 10:313–320
Maliro MFA, McNeil D, Kollmorgen J, Pittock C, Redden B (2004) Screening chickpea (Cicer arietinumL.) and wild relatives germplasm from diverse sources for salt tolerance. New directions for a diverse planet. In: Proceedings of the 4th International Crop Science Congress, Brisbane, Australia (September 26–October 1). Available at: http://www.cropscience.org.au
Maliro MFA, MacNeil D, Redden B, Kollmorgen JF, Pittock C (2008) Sampling strategies and screening of chickpea (Cicer arietinum L.) germplasm for salt tolerance. Genet Resour Crop Evol 55:53–63
Manchanda G, Garg N (2008) Salinity and its effects on the functional biology of legumes. Acta Physiol Plant 30:595–618
Manchanda HR, Sharma SK (1989) Tolerance of chloride and sulphate salinity in chickpea (Cicer arietinum). J Agric Sci 113::407–410
Mantri NL, Ford R, Coram TE, Pang EC (2007) Transcriptional profiling of chickpea genes differentially regulated in response to high-salinity, cold and drought. BMC Genom 8:303
Marconi G, Pace R, Traini A, Raggi L, Lutts S, Chiusano M, Guiducci M, Falcinelli M, Benincasa P, Albertini E (2013) Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS One 8:e75597
Massawe F, Mayes S, Cheng A (2016) Crop diversity: an unexploited treasure trove for food security. Trends Plant Sci 21:P365–P368
Merchan F, de Lorenzo L, Rizzo SG, Niebel A, Manyani H, Frugier F, Sousa C, Crespi M (2007) Identification of regulatory pathways involved in the reacquisition of root growth after salt stress in Medicago truncatula. Plant J 51:1–17
Mhadhbi H, Fotopoulos V, Mylona PV, Jebara M, Elarbi Aouani M, Polidoros AN (2011) Antioxidant gene-enzyme responses in Medicago truncatula genotypes with different degree of sensitivity to salinity. Physiol Plant 141:201–214
Mhadhbi H, Fotopoulos V, Mylona PV, Jebara M, Aouani ME, Polidoros AN (2013) Alternative oxidase 1 (Aox1) gene expression in roots of Medicago truncatula is a genotype-specific component of salt stress tolerance. J Plant Physiol 170:111–114
Moghaddam SM, Mamidi S, Osorno JM, Lee R, Brick M, Kelly J, Miklas P, Urrea C, Song Q, Cregan P, Grimwood J, Schmutz J, McClean PE (2016) Genome-wide association study identifies candidate loci underlying agronomic traits in a middle American diversity panel of common bean. Plant Genome. https://doi.org/10.3835/plantgenome2016.02.0012
Mokhtari F, Rafiei F, Shabani L, Shiran B (2017) Differential expression pattern of transcription factors across annual Medicago genotypes in response to salinity stress. Biol Plant 61:227–234
Molina C, Zaman-Allah M, Khan F, Fatnassi N, Horres R, Rotter B, Steinhauer D, Amenc L, Drevon JJ, Winter P, Kahl G (2011) The salt-responsive transcriptome of chickpea roots and nodules via deepSuperSAGE. BMC Plant Biol 11:31
Munns R, Gilliham M (2015) Salinity tolerance of crops—what is the cost? New Phytol 208:668–673
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59:651–681
Muñoz N, Ailin Liu A, Kan L, Li MW, Lam HM (2017) Potential uses of wild germplasms of grain legumes for crop improvement. Int J Mol Sci 18:328
Muñoz-Amatriaín M, Mirebrahim H, Xu P, Wanamaker SI, Luo M, Alhakami H, Alpert M, Atokple I, Batieno BJ, Boukar O, Bozdag S, Cisse N, Drabo I, Ehlers JD, Farmer A, Fatokun C, Gu YQ, Guo YN, Huynh BL, Jackson SA, Kusi F, Lawley CT, Lucas MR, Ma Y, Timko MP, Wu J, You F, Barkley NA, Roberts PA, Lonardi S, Close TJ (2017) Genome resources for climate-resilient cowpea, an essential crop for food security. Plant J 89:1042–1054
Murillo-Amador B, Troyo-Diéguez E, López-Cortés A, Jones HG, Ayala-Chairez F, Tinoco-Ojanguren CL (2001) Salt tolerance of cowpea genotypes in the emergence stage. Austra J Expt Agric 41:81–88
Murillo-Amador B, Troyo-Die´guez E, Garcı´a-Herna´ndez JL, Lo´pez-Aguilar R, A´vila-Serrano NY, Zamora-Salgado S, Rueda-Puente EO, Kaya C (2006) Effect of NaCl salinity in the genotypic variation of cowpea (Vigna unguiculata) during early vegetative growth. Sci Hort 108:423–431
Nabi F, Chaker-Haddadj A, Tellah S, Ghalem A, Ounane G, Ghalmi N, Djebbar R, Ounane SM (2017) Evaluation of Algerian cowpea genotypes for salt tolerance at germination stage. Adv Environ Biol 11:79–88
Najafi F, Khavari-Nejad RA, Rastgar-jazii F, Sticklen M (2006) Physiological changes in pea (Pisum sativum L. cv. Green Arrow) under NaCl salinity. Pak J Biol Sci 9:974–978
Najafi F, Khavari-Nejad RA, Rastgar-jazii F, Sticklen M (2007) Growth and some physiological attributes of pea (Pisum sativum L.) as affected by salinity. Pak J Biol Sci 10:2752–2755
Negrão S, Schmöckel SM, Tester M (2017) Evaluating physiological responses of plants to salinity stress. Ann Bot 119:1–11
Ngara R, Ndimba BK (2014) Understanding the complex nature of salinity and drought-stress response in cereals using proteomics technologies. Proteomics 14:611–621
Noreen Z, Ashraf M (2009) Assessment of variation in antioxidative defense system in salt-treated pea (Pisum sativum) cultivars and its putative use as salinity tolerance markers. J Plant Physiol 166:1764–1774
Özge Ç, Atak Ç (2012) Evaluation of proline accumulation and ∆1-pyrroline-5-carboxylate synthetase (P5CS) gene expression during salinity stress in two soybean (Glycine max L. Merr.) Varieties. Pol J Environ Stud 3:559–564
Pan WJ, Tao JJ, Cheng T, Bian XH, Wei W, Zhang WK, Ma B, Chen SY, Zhang JS (2016) Soybean miR172a improves salt tolerance and can function as a long-distance signal. Mol Plant 9:1337–1340
Pandey G, Sharma N, Sahu PP, Prasad M (2016a) Chromatin-based epigenetic regulation of plant abiotic stress response. Curr Genom 17:490–498
Pandey MK, Roorkiwal M, Singh VK, Ramalingam A, Kudapa H, Thudi M, Chitikineni A, Rathore A, Varshney RK (2016b) Emerging genomic tools for legume breeding: current status and future prospects. Front Plant Sci 7:455
Pandolfi C, Mancusoa S, Shabala S (2012) Physiology of acclimation to salinity stress in pea (Pisum sativum). Environ Expt Bot 84:44–51
Pantalone VR, Kenworthy WJ, Slauther LH, James BR (1997) Chloride tolerance in soybean and perennial Glycine a ccessions. Euphytica 97:235–239
Parida AK, Das AB (2005) Salt tolerance and salinity effects on plants: a review. Ecotoxicol Environ Saf 60:324–349
Patil G, Do T, Vuong TD, Valliyodan B, Lee JD, Chaudhary J, Shannon JG, Nguyen HT (2016) Genomic-assisted haplotype analysis and the development of high-throughput SNP markers for salinity tolerance in soybean. Sci Rep 6:19199
Paul S, Kundu A, Pal A (2011) Identification and validation of conserved microRNAs along with their differential expression in roots of Vigna unguiculata grown under salt stress. Plant Cell Tissue Organ Cult 105:233–242
Pennisi E (2013) The CRISPR craze. Science 341:833–836
Phang TH, Shao G, Lam HM (2008) Salt tolerance in soybean. J Integr Plant Biol 50:1196–1212
Pi E, Qu L, Hu J, Huang Y, Qiu L, Lu H, Jiang B, Liu C, Peng T, Zhao Y, Wang H, Tsai SN, Ngai S, Du L (2016) Mechanisms of soybean roots’ tolerances to salinity revealed by proteomic and phosphoproteomic comparisons between two cultivars. Mol Cell Proteom 15:266–288
Pushpavalli R, Krishnamurthy L, Thudi M, Gaur PM, Rao MV, Siddique KHM, Colmer TD, Turner NC, Varshney RK, Vadez V (2015) Two key genomic regions harbour QTLs for salinity tolerance in ICCV 2 × JG 11 derived chickpea (Cicer arietinum L.) recombinant inbred lines. BMC Plant Biol 15:124
Qi X, Li MW, Xie M, Liu X, Ni M, Shao G, Song C, Kay-Yuen Yim A, Tao Y, Wong FL, Isobe S, Wong CF, Wong KS, Xu C, Li C, Wang Y, Guan R, Sun F, Fan G, Xiao Z, Zhou F, Phang TH, Liu X, Tong SW, Chan TF, Yiu SM, Tabata S, Wang J, Xu X, Lam HM (2014) Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat Commun 5:4340
Qiu PC, Zhang WB, Liu CY, Jiang HW, Li CD, Fan HM, Zeng QL, Hu GH, Cheng QS (2011) QTL identification of salt tolerance in germination stage of soybean. Legume Genom Genet 2:20–27
Ramalingam A, Kudapa H, Pazhamala LT, Weckwerth W, Varshney (2015) Proteomics and Metabolomics: two emerging areas for legume Improvement. Front Plant Sci 6:1116
Rathi D, Gayen D, Gayali S, Chakraborty S, Chakraborty N (2016) Legume proteomics: progress, prospects, and challenges. Proteomics 16:310–327
Ravelombola W, Shi A, Weng Y, Mou B, Motes D, John Clark J, Chen P, Srivastava V, Qin J, Dong L, Yang W, Bhattarai G, Sugihara (2017) Association analysis of salt tolerance in cowpea (Vigna unguiculata (L.) Walp) at germination and seedling stages. Theor Appl Genet 131:79–91
Ren S, Weeda S, Li H, Whitehead B, Guo Y, Atalay A, Parry J (2012) Salt tolerance in soybean WF-7 is partially regulated by ABA and ROS signaling and involves withholding toxic Cl– ions from aerial tissues. Plant Cell Rep 31:1527–1533
Rendón-Anaya M, Montero-Vargas JM, Saburido-Álvarez S, Vlasova A, Capella-Gutierrez S, Ordaz-Ortiz JJ, Aguilar OM, Vianello-Brondani RP, Santalla M, Delaye L, Gabaldón T, Gepts P, Winkler R, Guigó R, Delgado-Salinas A, Herrera-Estrella A (2017) Genomic history of the origin and domestication of common bean unveils its closest sister species. Genome Biol 18:60
Roy SJ, Negrao S, Tester M (2014) Salt resistant crop plants. Curr Opin Biotechnol 26:115–124
Sahi C, Singh A, Kumar K, Blumwald E, Grover A (2006a) Salt stress response in rice: genetics, molecular biology, and comparative genomics. Funct Integr Genom 6:263–284
Sahi C, Singh A, Blumwald E, Grover A (2006b) Beyond osmolytes and transporters: novel plant salt stress tolerance-related genes from transcriptional profiling data. Physiol Plant 127:1–9
Salt DE, Baxter I, Lahner B (2008) Ionomics and the study of the plant ionome. Annu Rev Plant Biol 59:709–733
Samineni S, Siddique KHM, Gaur PM, Colmer TD (2011) Salt sensitivity of the vegetative and reproductive stages in chickpea (Cicer arietinum L.): Podding is a particularly sensitive stage. Environ Expt Bot 71:260–268
Sanchez DH, Siahpoosh MR, Roessner U, Udvardi M, Kopka J (2008) Plant metabolomics reveals conserved and divergent metabolic responses to salinity. Physiol Plant 132:209–219
Sanchez DH, Lippold F, Redestig H, Hannah MA, Erban A, Krämer U, Kopka J, Udvardi MK (2009) Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus. Plant J 53:973–987
Sanchez DH, Szymanski J, Erban A, Udvardi MK, Kopka J (2010) Mining for robust transcriptional and metabolic responses to long-term salt stress: a case study on the model legume Lotus japonicus. Plant Cell Environ 33:468–480
Sanchez DH, Pieckenstain FL, Escaray F, Erban A, Kraemer U, Udvardi MK, Kopka J (2011) Comparative ionomics and metabolomics in extremophile and glycophytic Lotus species under salt stress challenge the metabolic pre-adaptation hypothesis. Plant Cell Environ 34:605–617
Saxena AK, Rewari RB (1991) Influence of phosphate and zinc on growth, nodulation and mineral composition of chickpea (Cicer arietinum L.) under salt stress. World J Microbiol Biotech 7::202–205
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, Jenkins J, Shu S, Song Q, Chavarro C, Torres-Torres M, Geffroy V, Moghaddam SM, Gao D, Abernathy B, Barry K, Blair M, Brick MA, Chovatia M, Gepts P, Goodstein DM, Gonzales M, Hellsten U, Hyten DL, Jia G, Kelly JD, Kudrna D, Lee R, Richard MM, Miklas PN, Osorno JM, Rodrigues J, Thareau V, Urrea CA, Wang M, Yu Y, Zhang M, Wing RA, Cregan PB, Rokhsar DS, Jackson SA (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46:707–713
Serraj R, Krishnamurthy L, Upadhyaya HD (2004) Screening chickpea mini-core germplasm for tolerance to soil salinity. Int Chickpea Pigeonpea Newslett 11:29–32
Shabala S (2009) Salinity and programmed cell death: unravelling mechanisms for ion specific signalling. J Expt Bot 60:709–712
Shahid MA, Pervez MA, Balal RM, Abbas T, Ayyub CM, Mattson NS, Riaz A, Iqbal Z (2012a) Screening of pea (Pisum sativum L.) genotypes for salt tolerance based on early growth stage attributes and leaf inorganic osmolytes. Austra J Crop Sci 6:1324–1331
Shahid MA, Balal RM, Pervez MA, Abbas T, Ashfaq M, Ghazanfar U, Afzal M, Rashid A, Garcia-Sanchez F, Mattson NS (2012b) Differential response of pea (Pisum sativum L.) genotypes to salt stress in relation to the growth, physiological attributes antioxidant activity and organic solutes. Austra J Crop Sci 6:828–838
Shanon MC (1986) New insights in plant breeding efforts for improved salt tolerance. Hort Technol 6:96–99
Shao GH, Song JZ, Liu HL (1986) Preliminary studies on the evaluation of salt tolerance in soybean varieties. Acta Agron Sin 6:30–35
Shao GH, Wan CW, Chang RZ, ChenYW (1993) Preliminary study on the damage of plasma membrane caused by salt stress. Crops 1:39–40
Shao GH, Chang RZ, Chen YW, Yan SR (1994) Study on inheritance of salt tolerance in soybean. Acta Agron Sin 20:721–726
Sharifia M, Ghorbanlib M, Ebrahimzadeh H (2007) Improved growth of salinity-stressed soybean after inoculation with salt pre-treated mycorrhizal fungi. J Plant Physiol 164:1144–1151
Sharma S (2017) Prebreeding using wild species for genetic enhancement of grain legumes at ICRISAT. Crop Sci 57:1132–1144
Sharma SK, Kumar S (1990) Effect of salinization on growth and distribution of Na+ and Cl– in two genotypes of chickpea. Indian J Plant Physiol 33:269–274
Shelden MC, Roessner U (2013) Advances in functional genomics for investigating salinity stress tolerance mechanisms in cereals. Front Plant Sci 4:123
Shi X, Yan L, Yang C, Yan W, Moseley DO, Wang T, Liu B, Di R, Chen P, Zhang M (2018) Identification of a major quantitative trait locus underlying salt tolerance in ‘Jidou 12’ soybean cultivar. BMC Res Notes 11:95
Shulaev V, Cortes D, Miller G, Mittler R (2008) Metabolomics for plant stress response. Physiol Plant 132:199–208
Silveira JAG, Carvalho FEL (2016) Proteomics, photosynthesis and salt resistance in crops: an integrative view. J Proteomics 143:24–35
Singh AK (2004) The physiology of salt tolerance in four genotypes of chickpea during germination. J Agric Sci Technol 6:87–93
Singh AK, Ganapathysubramanian B, Sarkar S, Singh A (2018) Deep learning for plant stress phenotyping: trends and future perspectives. Trends Plant Sci 23:883–898
Slemi N, Lachaal M, Andelly C, Soltani A, Hajji M (2001) Physiological behaviour of two chickpea Tunisian varieties irrigated with saline nutrient solution. Dev Plant Soil Sci 92:408–409
Sobhanian H, Razavizadeh R, Nanjo Y, Ehsanpour AA, Jazii FR, Motamed N, Setsuko K (2010) Proteome analysis of soybean leaves, hypocotyls and roots under salt stress. Proteome Sci 8:19
Song Y, Ji D, Li S, Wang P, Li Q, Xiang F (2012) The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS One 7:e41274
Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, Cregan PB (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One 8:e54985
Sumner LW, Mendes P, Dixon RA (2003) Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochem 62:817–836
Sun YX, Wang D, Bai YL, Wang NN, Wang Y (2006) Studies on the over expression of the soybean GmNHX1 in Lotus corniculatus: the reduced Na+ level is the basis of the increased salt tolerance. Chin Sci Bull 51:1306–1315
Tanksley SD, McCouch SR (1997) Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277:1063–1066
Tejera NA, Soussi M, Lluch C (2006) Physiological and nutritional indicators of tolerance to salinity in chickpea plants growing under symbiotic conditions. Environ Expt Bot 58:17–24
Thiam M, Champion A, Diouf D, Ourèye SY M (2013) NaCl effects on in vitro germination and growth of some Senegalese cowpea (Vigna unguiculata (L.) Walp.) cultivars. ISRN Biotechnology 2013:11
Tsai SN (2003) Cloning and characterization of ion transporters genes from a salt-tolerant soybean variety. M.Phil. Thesis. The Chinese University of Hong Kong
Turner NC, Colmer TD, Quealy J, Pushpavalli R, Krishnamurthy L, Kaur J, Singh G, Siddique KHM, Vadez V (2013) Salinity tolerance and ion accumulation in chickpea (Cicer arietinum L.) subjected to salt stress. Plant Soil 365:347–361
Tuyen DD, Lal SK, Xu DH (2010) Identification of a major QTL allele from wild soybean (Glycine soja Sieb. & Zucc.) for increasing alkaline salt tolerance in soybean. Theor Appl Genet 121:229–236
Tuyen DD, Zhang HM, Xu DH (2013) Validation and high-resolution mapping of a major quantitative trait locus for alkaline salt tolerance in soybean using residual heterozygous line. Mol Breed 31:79–86
Umezawa T, Shimizu K, Kato M, Ueda T (2000) Enhancement of salt tolerance in soybean with NaCl pretreatment. Physiol Plant 110:59–66
Vadez V, Krishnamurthy L, Serraj R, Gaur PM, Upadhyaya HD, Hoisington DA, Varshney RK, Turner NC, Siddique KHM (2007) Large variation in salinity tolerance in chickpea is explained by differences in sensitivity at the reproductive stage. Field Crops Res 104:123–129
Vadez V, Rashmi M, Sindhu K, Muralidharan M, Pushpavalli R, Turner NC, Krishnamurthy L, Gaur PM, Colmer TD (2012a) Large number of flowers and tertiary branches, and higher reproductive success increase yields under salt stress in chickpea. Eur J Agron 41:42–51
Vadez V, Krishnamurthy L, Thudi M, Anuradha C, Colmer TD, Turner NC, Siddique KHM, Gaur PM, Varshney RK (2012b) Assessment of ICCV 2 9 JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTL for seed yield and yield components. Mol Breed 30:9–21
Valencia R, Chen P, Ishibashi T, Conatser M (2008) A rapid and effective method for screening salt tolerance in soybean. Crop Sci 48:1773–1779
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KH, Town CD, Hoisington DA (2009) A comprehensive resource of drought- and salinity- responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genom 10:523
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Verma M, Kumar V, Patel RK, Garg R, Jain M (2015) CTDB: an integrated chickpea transcriptome database for functional and applied genomics. PLoS One 10:e0136880
Vu WT, Chang PL, Moriuchi KS, Friesen ML (2015) Genetic variation of transgenerational plasticity of offspring germination in response to salinity stress and the seed transcriptome of Medicago truncatula. BMC Evol Biol 15:59
Wang D, Shannon MC (1999) Emergence and seedling growth of soybean cultivars and maturity groups under salinity. Plant Soil 214:117–124
Wang W, Vinocur B, Altman A (2003) Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218:1–14
Wang M, Qin L, Xie C, Li W, Yuan J, Kong L, Yu W, Xia G, Liu S (2014) Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol 55:1354–1365
Wang W, Huang F, Qin Q, Zhao X, Li Z, Fu B (2015) Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem Biophys Res Commun 465:790–796
Wang C, Hu S, Gardner C, Lubberstedt T (2017) Emerging avenues for utilization of exotic germplasm. Trends Plant Sci 22:624–637
Wang Y, Jiang L, Chen J, Tao L, An Y, Cai H, Guo C (2018) Over expression of the alfalfa WRKY11 gene enhances salt tolerance in soybean. PLoS One 13:e0192382
Win KT, Oo AZ (2015) Genotypic difference in salinity tolerance during early vegetative growth of cowpea (Vigna unguiculata L. Walp.) from Myanmar. Biocat Agril Biotechnol 4:449–455
Wong TH, Li MW, Yao XQ, Lam HM (2013) The GmCLC1 protein from soybean functions as a chloride ion transporter. J Plant Physiol 170:101–104
Wu YB, Yu BJ (2009) Gene expression of Cl) channel and its relation to chloride tolerance of Glycine max, Glycine soja and their hybrid seedlings. J Nanjing Agri Univ 32:67–71 (in Chinese)
Wu D, Shen Q, Cai S, Chen ZH, Dai F, Zhang G (2013) Ionomic responses and correlations between elements and metabolites under salt stress in wild and cultivated barley. Plant Cell Physiol 54:1976–1988
Wu G, Zhou Z, Chen P, Tang X, Shao H, Wang H (2014) Comparative eco physiological study of salt stress for wild and cultivated soybean species from the Yellow River Delta, China. Sci World J 2014:651745
Xu D, Tuyen D (2010) Genetic studies on saline and sodic tolerances in soybean. Breed Sci 61:559–565
Xu P, Wu X, Muñoz-Amatriaín M, Wang B, Wu X, Hu Y, Huynh BL, Close TJ, Roberts PA, Zhou W, Lu Z, Li G (2017) Genomic regions, cellular components and gene regulatory basis underlying pod length variations in cowpea (V. unguiculata L. Walp). Plant Biotechnol J 15:547–557
Yang J (2008) Development and prospect of the research on salt-affected soils in China. Acta Pedol Sin 45:837–845 (in Chinese)
Yasuta Y, Kokubun M (2014) Salinity tolerance of super-nodulating soybean Genotype En-b0-1. Plant Prod Sci 17:32–40
Yu BJ, Lam HM, Shao GH, Liu YL (2005) Effects of salinity on activities of H+-ATPase, H+-PPase and membrane lipid composition in plasma membrane and tonoplast vesicles isolated from soybean (Glycine max L.) seedlings. J Environ Sci 17:259–262
Yu Y, Wang N, Hu R, Xiang F (2016) Genome–wide identification of soybean WRKY transcription factors in response to salt stress. Springer Plus 5:920
Zahaf O, Blanchet S, de Zélicourt A, Alunni B, Plet J, Laffont C, de Lorenzo L, Imbeaud S, Ichanté JL, Diet A, Badri M, Zabalza A, González EM, Delacroix H, Gruber V, Frugier F, Crespi M (2012) Comparative transcriptomic analysis of salt adaptation in roots of contrasting Medicago truncatula genotypes. Mol Plant 5:1068–1081
Zargar SM, Mahajan R, Nazir M, Nagar P, Kim ST, Rai V, Masi A, Ahmad SM, Shah RA, Ganai NA, Agrawal GK, Rakwal R (2017) Common bean proteomics: Present status and future strategies. J Proteom S1874-3919:30104–30105
Zeng A, Chen P, Korth K, Hancock F, Pereira A, Brye K, Wu C, Shi A (2017) Genome-wide association study (GWAS) of salt tolerance in worldwide soybean germplasm lines. Mol Breed 37:30
Zeng A, Chen P, Korth KL, Ping J, Thomas J, Wu C, Srivastava S, Pereira A, Hancock F, Brye K, Ma J (2018) RNA sequencing analysis of salt tolerance in soybean (Glycine max). Genomics 4(18)30196
Zhang XK, Zhou QH, Cao JH, Yu BJ (2011) Differential Cl)/salt tolerance and NaCl-induced alternations of tissue and cellular ion fluxes in Glycine max, Glycine soja and their Hybrid Seedlings. J Agron Crop Sci 197:329–339
Zhang WJ, Niu Y, Bu SH, Li M, Feng JY, Zhang J, Yang SX, Odinga MM, Wei SP, Liu XF, Zhang YM (2014) Epistatic association mapping for alkaline and salinity tolerance traits in the soybean germination stage. PLoS One 9:e84750
Zhang J, Yang D, Li M, Shi L (2016) Metabolic profiles reveal changes in wild and cultivated soybean seedling leaves under salt stress. PLoS One 11:e0159622
Zhao SP, Lu D, Yu TF, Ji YJ, Zheng WJ, Zhang SX, Chai SC, Chen ZY, Cui XY (2017) Genome-wide analysis of the YABBY family in soybean and functional identification of GmYABBY10 involvement in high salt and drought stresses. Plant Physiol Biochem 119:132–146
Zhou Z, Jiang Y, Wang Z, Gou Z, Lyu J, Li W, Yu Y, Shu L, Zhao Y, Ma Y, Fang C, Shen Y, Liu T, Li C, Li Q, Wu M, Wang M, Wu Y, Dong Y, Wan W, Wang X, Ding Z, Gao Y, Xiang H, Zhu B, Lee SH, Wang W, Tian Z (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 33:408–414
Zhu H, Choi H, Cook DR, Shoemaker RC (2005) Bridging model and crop legumes through comparative genomics. Plant Physiol 137:1189–1196
Acknowledgements
The authors acknowledge support from Indian Council of Agricultural Research (ICAR), India.
Funding
The authors have not received any funding for writing this MS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by Bettina Hahne.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jha, U.C., Bohra, A., Jha, R. et al. Salinity stress response and ‘omics’ approaches for improving salinity stress tolerance in major grain legumes. Plant Cell Rep 38, 255–277 (2019). https://doi.org/10.1007/s00299-019-02374-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-019-02374-5