
Abstract
Bacterial non-specific nucleases of the phospholipase D family are widely distributed among the members of the Enterobacteriaceae. Each genome mainly contains a single copy of a gene encoding a phospholipase D family protein. However, two distantly related isozymes (< 40% identity at the protein level) were identified by BLAST-analyses in the plant pathogenic competitor enterobacterium Pantoea agglomerans. The two nucleases PaNuc-1 and PaNuc-2 were produced in Escherichia coli. Identical gene constructs and expression conditions resulted in the production of PaNuc-1 in soluble form, while PaNuc-2 remained insoluble in inclusion bodies. PaNuc-2 was refolded and both proteins were purified by a combination of affinity and ion exchange chromatography. Proteolytic removal of the HIS-tag allowed the characterization of pure and mature tag-less proteins. Enzymatic properties of both isozymes revealed that they are non-specific nucleases, displaying activities against RNA, single- and double-stranded genomic DNA as well as circular plasmids. However, their biochemical activity profiles were clearly different, with PaNuc-1 being optimally active at 70 °C and pH 7.0, while PaNuc-2 was most active at 45 °C and pH 7.0. The enzymes retained > 90% nuclease activity at EDTA concentrations of 4 mM (PaNuc-2) and 20 mM (PaNuc-1), respectively. Different enzymatic properties suggest that the roles of PaNuc-1 and PaNuc-2 differ in the cell and might be the result of functional diversification after an ancient gene duplication event took place. The fact that both enzymes could be easily produced in recombinant form and their tolerance against metal ion chelators in combination with a broad substrate promiscuity might pave the way to versatile biotechnological applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The group of nucleases is ubiquitously distributed among all organisms and cells and fulfills versatile functionalities (Yang 2011). Intracellular nucleases are involved in cellular processes such as repair and restriction of nucleic acids, in recombination approaches or replication processes, while the decay of nucleic acids as a cause of cell death is probably the main task of extracellular non-specific nucleases (NSN).
NSN are classified based on their mode of action, choice of substrates and nature of reaction products. Isozymes may act on different types of nucleic acids (DNA or RNA), produce mono- or oligonucleotides as major reaction products, and catalyze the hydrolysis of nucleic acids in an exo- or endo-acting manner (Rangarajan and Shankar 2001). Some are also able to distinguish between nucleic acid structures. NSN are capable of catalyzing the hydrolysis of nucleic acids independently of a certain sequence and can be precisely distinguished from the routinely used restriction endonucleases that recognize a specific sequence prior to its hydrolysis. Typical nucleases are characterized by a divalent metal-ion in their catalytic region (mostly Mg2+), which is essential for enzymatic activity (Li and Rohrmann 2000).
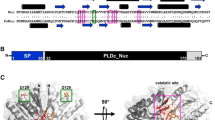
Enzymes of the phospholipase D (PLD) family (EC 3.1.4.4) were initially described to hydrolyze phosphatidylcholine and were found in mammals and plants. Nowadays, the superfamily of proteins exhibiting PLD-motifs comprises mammalian and plant enzymes such as cardiolipin synthases and phosphatidylserine synthases as well as structural proteins including the poxvirus envelope protein or a murine toxin from Yersinia pestis (Leiros et al. 2000; Ponting and Kerr 1996; Rudolph et al. 1999). In 1997, a related bacterial endonuclease from Salmonella enterica subsp. enterica serovar Typhimurium (formerly known as Salmonella typhimurium) was characterized and was also assigned to the group of PLD proteins (Zhao et al. 1997). A highly conserved HxK(x)4D(x)6GSxN motif is duplicated in nearly all mammalian PLD proteins, while homologs of the bacterial endonuclease exhibit only a single copy of the motif (Stuckey and Dixon 1999) (Fig. 1). This nucleic acid–degrading enzyme acts in a non-specific manner and can be distinguished from other nucleases due to its metal-ion independent catalytic activity. So far, only a few nucleases that are not dependent on the presence of metal-ions have been characterized (Bao et al. 2008; Grazulis et al. 2005; Song and Zhang 2008; Stuckey and Dixon 1999; Yang 2011).

Parallel sequence alignment of PaNuc-1 (WP_039390287.1) and PaNuc-2 (WP_033781266.1). The alignment was generated with ClustalX. Conserved amino acid residues of the HxK(x)4D(x)6GSxN are boxed in green, while the Asp119Gly mutation in PaNuc-2 is highlighted by a red box. Predicted signal peptide cleavage sites are indicated by black arrows
NSN are of industrial importance because they can be applied to remove nucleic acids in downstream processing approaches. A commercially available nuclease from Serratia marcescens (trademark “Benzonase® Nuclease”) is widely used for the elimination of nucleic acid contaminations during protein purification and single cell protein preparations, while bovine DNase I is not active towards RNA and can be used for DNA digestion during RNA purification. Such enzymes have also been extensively used as analytical tools to determine structures of nucleic acids (Rangarajan and Shankar 2001). Moreover, metal-ion-independent nucleases are promising candidates for the application in downstream processing to reduce the viscosity of crude protein extracts in EDTA-containing buffer solutions (Schmitz et al. 2019).
Pantoea agglomerans is a Gram-negative, aerobic bacterium of the family Enterobactericeae that is commonly distributed in plants and soil. It is an opportunistic pathogen infecting wounds and the urinary tract in patients (Cruz et al. 2007). In this study, two distantly related putative proteins of the PLD family encoded in the genome of P. agglomerans were retrieved from public databases and produced in recombinant form in E. coli. After proteolytic cleavage of the HIS-tagged precursor proteins, mature enzymes were comparably characterized in detail and evolutionary aspects as well as enzymatic properties are reported.
Material and methods
Strains and culture conditions
Escherichia coli NEB® 5-alpha (New England Biolabs, Frankfurt/Main, Germany) was used for plasmid maintenance and propagation. E. coli Veggie BL21 (DE3) (New England Biolabs, Frankfurt/Main, Germany) was used for gene expression and protein production. Expression strains were either grown in Luria-Bertani (10 g/L NaCl, 5 g/L Veggie Yeast Extract, 10 g/L Veggie Peptone) medium for cloning purposes or in 5× Yeast-Peptone (25 g/L Veggie Yeast Extract, 16 g/L peptone from soybean, 5 g/L NaCl) medium for high-cell-density fermentation approaches.
Gene analyses
BLASTP analyses were applied to screen for bacterial nucleases. Protein sequence data of putative and characterized bacterial PLD-family proteins were obtained from the public databases UNIPROT (http://www.uniprot.org/) and NCBI (https://www.ncbi.nlm.nih.gov/pubmed/). Multiple protein sequence alignments were generated with ClustalX version 2.1. Pairs of distantly related isozymes exhibiting identities between 15 and 40% on the amino acid level were selected from individual bacterial genomes to discover nucleases with varying biochemical properties resulting from diversification processes in the course of evolution. Conserved protein domains were identified with the Conserved Domain Database (CDD), and signal peptide predictions were done with SignalP version 4.1 (Larkin et al. 2007; Marchler-Bauer et al. 2015; Nielsen 2017).
Cloning of the genes encoding nucleases
The genes encoding two putative nucleases (WP_039390287.1 – PaNuc-1; WP_033781266.1 – PaNuc-2) from the Gram-negative enterobacterium Pantoea agglomerans were codon-optimized and synthesized at ATUM (Newark, California, USA). Synthetic genes were provided in subcloning vector pJAmp and recloned in frame with an N-terminal tandem HIS (2HISLinker) fusion encoding DNA sequence into pET24d(+) (Merck Chemicals GmbH, Darmstadt, Germany) expression vector for cytosolic protein production. The derived plasmids pET24d::PaNuc-1 and pET24d::PaNuc-2 were used to transform E. coli Veggie BL21 (DE3) to produce HIS-tagged nuclease variants.
Recombinant expression
E. coli expression strains harboring expression plasmids were grown in a 1.2-L fed-batch fermentation at 30 °C in 5× Yeast-Peptone medium supplemented with 50 μg/ml kanamycin with 1200 rpm max. After an optical density at 600 nm of OD600 = 40–45 was reached, temperature was increased to 37 °C and gene expression was induced using a final concentration of 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Cells were harvested by centrifugation after 4 h at an OD600 = 50–60.
Purification of soluble nuclease
Pelleted cells were dissolved in 20 mM NaH2PO4, pH 8.0 (5 ml buffer per gram wet weight E. coli cells) and disrupted by sonication. Cell debris was precipitated by centrifugation (20,035g, 30 min, 4 °C) and the cleared lysate was loaded on a polypropylene column equipped with Ni-Sepharose (GE Healthcare, Munich, Germany) that was equilibrated with 50 mM NaH2PO4, 50 mM NaCl, 5 mM imidazole, pH 7.0. The column was washed with 50 mM NaH2PO4, 50 mM NaCl, 50 mM imidazole, pH 7.0 and subsequently with 50 mM NaH2PO4, 50 mM NaCl, 150 mM imidazole, pH 7.0. 50 mM NaH2PO4, 50 mM NaCl, 500 mM imidazole, pH 7.0 was used for elution, nuclease-positive fractions were pooled, diluted at least 1:4 with 25 mM NaH2PO4, pH 6.0 until the conductivity was < 9 mS/cm, and loaded onto a column equipped with SP-sepharose (GE Healthcare, Munich, Germany) equilibrated with 25 mM NaH2PO4, pH 6.0. Column was washed with 25 mM NaH2PO4, 300 mM NaCl, pH 6.0 and the target protein was eluted with 25 mM NaH2PO4, 1 M NaCl, pH 6.0. Afterwards, purified 2HIS-PaNuc-1 was incubated with one unit protease overnight at 4 °C. The enzymatically processed protein fraction was loaded on Ni-Sepharose equilibrated with 25 mM NaPO4, 25 mM NaCl, pH 7.0 to separate PaNuc-1 in the flow-through fraction from 2HISLinker-tag and HIS-tagged protease. Finally, buffer exchange was achieved using PD10 desalting columns (GE Healthcare, Munich, Germany) to store pure PaNuc-1 in 25 mM NaPO4, 25 mM NaCl, pH 7.0 at 4 °C.
Purification of insoluble nuclease from inclusion bodies
For the purification of insoluble nuclease, pelleted cells were resuspended in 20 mM NaH2PO4, 1 mM MgCl2, 1 mM beta-mercaptoethanol, pH 8.0 (5 ml buffer per gram wet weight E. coli cells) and disrupted by sonication. Crude extracts were incubated at 37 °C for 30 min and then centrifuged (20,035g, 30 min, 4 °C). Supernatant was discarded and the insoluble protein fraction including cell debris (inclusion bodies: IBs) was resuspended in 30 ml IB-washing buffer 1 (50 mM NaH2PO4, 100 mM NaCl, 1% (v/v) Triton X-100, 5 mM Ethylenediaminetetraacetic acid (EDTA), 1 mM beta-mercaptoethanol, pH 8.0) per gram of pellet. Solubilized IBs were sonicated and rotated at 20 °C for 10 min on a MACSmix™ (Miltenyi Biotec GmbH, Germany). After centrifugation (5195g, 20 min, 4 °C), supernatant was discarded and IBs were resolubilized in IB-washing buffer 1. The washing process was repeated three times before resuspension in 100 ml IB-washing buffer 2 (PBS, 5 mM EDTA, 1 mM beta-mercaptoethanol, pH 7.5) per gram of pellet took place. IBs were sonicated and sedimented (5195g, 20 min, 4 °C) again followed by solubilization in 20 mM NaH2PO4, 6 M guanidine-HCl, 1 mM beta-mercaptoethanol, pH 8.0. A final sonication step was applied and resuspended IBs were incubated at 4 °C for 24 h on a MACSmix™ (Miltenyi Biotec GmbH, Germany) to guarantee complete solubilization. Remaining debris was sedimented by centrifugation (6795g, 20 min, 20 °C) and the supernatant containing solubilized IBs was collected and stored at − 20 °C. Refolding of washed IBs was conducted by rapid dilution of 1:50 in 20 mM NaH2PO4, 500 mM l-arginine, 1 mM beta-mercaptoethanol, pH 8.5. l-Arginine was applied as an additive to promote refolding of the solubilized nuclease and to prevent aggregation (Singh et al. 2015). Protein solution was constantly stirred for 3 days at 4 °C. Finally, proteins were diluted 1:10 in 20 mM NaH2PO4, 1 mM beta-mercaptoethanol, pH 8.5 and constantly stirred for further 24 h at 4 °C. The protein solution was loaded onto a column equipped with Q-sepharose (GE Healthcare, Munich, Germany) that was equilibrated with 25 mM NaH2PO4, 1 mM beta-mercaptoethanol, pH 8.5. The column was washed with 25 mM NaH2PO4, 1 mM beta-mercaptoethanol, 50 mM NaCl, pH 8.5 and the target protein was eluted with 25 mM NaH2PO4, 1 mM beta-mercaptoethanol, 1 M NaCl, pH 8.5. PaNuc-2–containing fractions were diluted 1:4 with 50 mM NaH2PO4, 10 mM imidazole, pH 8.0 and loaded on a polypropylene column equipped with Ni-Sepharose (GE-Healthcare, Munich, Germany) equilibrated with 50 mM NaH2PO4, 50 mM NaCl, 5 mM imidazole, pH 8.0. After washing the column with 50 mM NaH2PO4, 50 mM NaCl, 25 mM imidazole, pH 8.0, the protein was eluted with 50 mM NaH2PO4, 50 mM NaCl, 250 mM imidazole, pH 8.0. Pure 2HIS-PaNuc-2 was incubated with one unit protease overnight at 4 °C. The enzymatically processed protein fraction was loaded on Ni-Sepharose equilibrated with 25 mM NaPO4, 25 mM NaCl, pH 7.0 to separate PaNuc-2 in the flow-through from 2HISLinker-tag and HIS-tagged protease. Finally, buffer exchange was achieved using PD10 desalting columns (GE Healthcare, Munich, Germany) to store pure PaNuc-2 in 25 mM NaPO4, 25 mM NaCl, pH 7.0 at 4 °C.
SDS-PAGE and Western blotting analyses
Visualization of heterologously produced nucleases was achieved by SDS-PAGE (ProGel Tris Glycin 4–20%, anamed Elektrophorese GmbH, Groß-Bieberau/Rodau, Germany) in an X Cell SureLock™ Mini-Cell Electrophoresis System (Thermo Fisher Scientific, Darmstadt, Germany). Protein transfer to Nitrocellulose blotting membrane (GE-Healthcare, Munich, Germany) was achieved using the semi-dry Western blotting system (Biometra, Göttingen, Germany). The detection of HIS-tagged nucleases was carried out with an anti-HIS HRP antibody (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) in combination with the Immobilon™ Western HRP substrate (Merck, Darmstadt, Germany). Mass spectrometry methods were used to accurately determine molecular masses of intact proteins with the micrOTOF-Q II Benchtop Mass Spectrometer (Bruker, Billerica, USA). Reduced (supplemented with 1 mM dithiothreitol) and non-reduced protein samples were analyzed to identify the formation of intramolecular disulfide bridges.
Enzyme activity assays
The activity of nucleases was determined either by a qualitative or by a quantitative assay. To perform a qualitative assay, about 100–200 ng of purified enzyme was incubated for 30 min in reaction buffer supplemented with DNA at a concentration of 5 μg in a final volume of 20 μl. Incubation took place for 10 min at 35 °C. Protein concentrations were determined by using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Darmstadt, Germany). Reaction was stopped by the addition of 0.25% (w/v) sodium dodecyl sulfate (SDS). Samples were supplemented with loading dye (Thermo Fisher Scientific, Darmstadt, Germany) and loaded onto a 1% (v/v) agarose gel. Running the gel took place in 1× TAE-buffer (Tris-acetate-EDTA), at a constant electric voltage of 100 V for 60 min to separate nucleic acids according to their sizes. Nucleic acids were stained with ethidium bromide. Concentrations of metal ions and chelators were varied to characterize enzymatic properties and stabilities. Substrate specificity was tested using circularized plasmid DNA, total RNA, single-stranded DNA (ssDNA) from calf thymus (Sigma-Aldrich, St. Louis, USA), and double-stranded sheared and unsheared genomic DNA (dsDNA) namely UltraPure™ Salmon Sperm DNA Solution (Thermo Fisher Scientific, Darmstadt, Germany) and Deoxyribonucleic acid from calf thymus (Sigma-Aldrich, St. Louis, USA).
In quantitative activity assays, about 10 ng of purified enzyme was incubated for 30 min in reaction buffer supplemented with DNA at a concentration of 0.5 μg in a final volume of 100 μl. Reaction was stopped using one volume of 4% (v/v) perchloric acid. Samples were thoroughly vortexed and incubated on ice for 45 min prior to centrifugation at 4 °C, 10 min, 15,115g. The VICTOR™ X4 Multilabel Plate Reader (PerkinElmer, Rodgau, Germany) in combination with Corning® 96-well UV-transparent plates (Merck Chemicals GmbH, Darmstadt, Germany) was used to determine enzymatic activities. Influence of chemical components on nucleases was investigated by pre-incubating substrate-free samples on ice for 60 min prior to activity assays under standard conditions. Reactions were started when reaction temperature was reached by the addition of sheared dsDNA.
Enzymatic kinetics were determined using sheared dsDNA in the following concentrations: 15 μM, 30 μM, 75 μM, 150 μM, 300 μM, 600 μM, 1.2 mM, and 1.8 mM, respectively. To calculate micromoles from concentrations of sheared dsDNA given in micrograms per milliliter, a mean molar mass per nucleotide of 330 g/mol was used as described earlier (MacLellan and Forsberg 2001). One unit of nuclease activity is defined as ΔA260 min−1 per milligram protein, while Kcat values were calculated from Vmax by the assumption that the degradation of 0.05 mg DNA resulted in a change in absorbance of 0.3 as described previously (MacLellan and Forsberg 2001). Kcat was obtained by using the general formula Kcat = Vmax/[E0].
The Amplex® Red Phospholipase D Assay Kit (Thermo Fisher Scientific, Pinneberg, Germany) was used to determine potential phospholipase D activity of nucleases from P. agglomerans. Phospholipase D from Streptomyces chromofuscus (Merck Chemicals GmbH, Darmstadt, Germany) was used as a positive control. Assays were conducted as described by the manufacturers.
Sequence accession numbers
The codon-optimized sequences of PaNuc-1 and PaNuc-2 from P. agglomerans were deposited under accession numbers LS998013 and LS998014 in the EMBL database.
Results
Identification of two putative nucleases from Pantoea agglomerans
A genome screening approach for bacterial non-specific nucleases that are not dependent on metal-ions was done using the nuclease domains of site-specific restriction endonucleases BfiI from Bacillus firmus and BmrI from Bacillus megaterium as well as the full-length endonuclease Nuc from Salmonella enterica subsp. enterica serovar Typhimurium as target sequences. These enzymes are characterized by the presence of the conserved HxK(x)4D(x)6GSxN motif. BLASTP analyses resulted in the identification of multiple hits including two open reading frames in the genome of the plant pathogen competitor bacterium Pantoea agglomerans. The deduced proteins were annotated as “phospholipase D family protein” (WP_039390287.1) and “endonuclease” (WP_033781266.1), respectively. Alignments of deduced amino acid residues of both protein sequences resulted in an identity of 39.5% in 167 aa overlap. The protein sequences exhibited lengths of 178 and 173 amino acid residues, including predicted N-terminal signal peptides of 22 and 20 amino acids (SignalP 4.1 Server, (Petersen et al. 2011)). The predicted molecular mass and the isoelectric point pI of PaNuc-1 are 19.3 kDa and 9.3, while PaNuc-2 is characterized by a molecular mass of 18.9 kDa and a pI of 6.9 (Table 1). Conserved domain searches revealed the presence of putative PLDc-Nuc domains in both proteins that belong to the family of phospholipase D and comprise a catalytic domain known from the EDTA-resistant nuclease Nuc (PDB: 1BYS_A). Amino acid alignments of Nuc, PaNuc-1, and PaNuc-2; and the bacterial restriction enzymes BfiI and BmrI; and the homolog from the EDTA-degrading bacterium Chelativorans multitrophicus revealed the identification of the conserved amino acids His105 (in BfiI; His117 and His111 in PaNuc-1 and PaNuc-2, respectively), Lys107, Asn125, and Glu136 that are essential for catalytic activity of BfiI. In addition, the HxK(x)4D(x)6GSxN motif is conserved in PaNuc-1, while Asp119 is replaced by Gly119 in PaNuc-2 (Fig. 1).
Production of the recombinant enzymes in Escherichia coli
Bacterial nucleases were inserted into pET24d-expression vector to produce recombinant 2HIS-fusion enzymes (2HISLinker-tag) in Escherichia coli Veggie BL21 (DE3) as described in “Materials and Methods” (Fig. 2A). Clones harboring the plasmid pET24d::PaNuc-1 exhibited a transparent colony phenotype and displayed a decreased growth velocity in liquid culture, while E. coli Veggie BL21 (DE3) transformed with plasmid pET24d::PaNuc-2 showed normal growth on solid agar plates (Fig. S1) and in liquid cultures (data not shown). Isolation of crude protein extracts revealed that the expression of the gene encoding PaNuc-1 resulted in soluble recombinant protein. Efficient purification using a combined approach of affinity and ionic exchange chromatography was established, prior to proteolytic cleavage to remove the tag (Fig. 2B). In contrast to these results, purification of PaNuc-2 demanded a more complex strategy starting with the recovery of the nuclease from unfolded inclusion bodies. Afterwards, a strategy composed of ion exchange chromatography as the first step, followed by affinity chromatography and tag removal, was applied (Fig. 2C). Incubation of PaNuc-1 in ion exchange elution buffer (25 mM NaH2PO4, 1 M NaCl) at pH 6.0 resulted in spontaneous cleavage of the 2HISLinker-tag, while refolded PaNuc-2 precursor eluted in affinity chromatography buffer (50 mM NaH2PO4, 50 mM NaCl, 250 mM imidazole) at pH 8.0 remained unprocessed until specific tag removal (Fig. 2) as a result of proteolytic cleavage took place. SDS-PAGE and Western blot revealed protein bands with an apparent molecular weight of 16 kDa each, which is consistent with the calculated molecular weights of the cleaved monomers (PaNuc-1: 16,990 Da; PaNuc-2: 16,567 Da). Determination of intact molecular masses also confirmed the molecular weight of PaNuc-1 (experimental data: 16,990 Da, same result for reduced and oxidized form, respectively). Reduced PaNuc-2 exhibited a molecular weight of 16,568 Da, while oxidized PaNuc-2 displayed a molecular weight of 16,566 Da, indicating the formation of a single disulfide bond in the enzyme. This result is in good agreement with the presence of two cysteines (Cys96 and Cys170) in mature PaNuc-2, while PaNuc-1 does not contain any cysteine (Table 1). Both proteins were purified with a yield of 23% and 10%, and a final hydrolytic activity towards dsDNA of 405 and 830 U/mg, respectively (Table S1).

Expression of nuclease-encoding genes in E. coli. SDS-PAGE and Western blotting analysis of E. coli protein extracts and purification samples. A Schematic illustration of the fusion enzyme construct composed of a twin HIS tag, linker and the bacterial nuclease. The cleavage site is indicated by a triangle. Molecular weight of the 2HISLinker-moiety (12.3 kDa) and of tag-free nucleases (PaNuc-1: 17.0 kDa and PaNuc-2: 16.6 kDa) are indicated below the graphical illustration. Expression strains either harbor plasmid (B) pET24d::PaNuc-1 or (C) pET24d::PaNuc-2, respectively. Total cellular proteins of overnight growth cultures (ON) or of induced (+IPTG) and non-induced cultivations (-IPTG) were resolved by SDS-PAGE and visualized by Coomassie Blue staining. Insoluble (PE pellet fraction) and soluble (SN supernatant) protein fractions after cell disruption were separated by centrifugation. Soluble proteins or washed and refolded inclusion bodies (IB) were purified by affinity (Affinity) and ion exchange (IEX) chromatography. Cleavage products after tag removal were visualized by SDS-PAGE and Western blot prior (Cleavage) and after final affinity purification using an anti-HIS antibody (PaNuc-1, PaNuc-2)
Biochemical properties of non-specific nucleases
The substrate specificity of recombinant nucleases was tested using phosphatidylcholine (data not shown), RNA, sheared and unsheared genomic dsDNA, and plasmid DNA and ssDNA (Fig. 3). PaNuc-1 and PaNuc-2 were both active on all types of nucleic acids tested, but did not cleave phosphatidylcholine to give phosphatidic acid and choline. Temperature and pH optima and activity ranges were determined in a quantitative assay using sheared dsDNA as substrate. PaNuc-1 and PaNuc-2 displayed temperature optima of 70 °C and 45 °C, respectively (Fig. 4A, B). Moreover, PaNuc-1 is optimally active at a pH range between 7.0 and 8.0, while the residual activity at pH 9.0 is reduced to 42.4 ± 1.3% and at pH 6.0 to 65.1 ± 1.3%, respectively (Fig. 5A). In contrast to these results, PaNuc-2 displayed > 50% activity in a broad pH range, with an optimum at pH 6.0 to pH 7.0 (Fig. 5B). Enzyme stabilities were tested with regard to destabilizing effects of increasing temperatures and different pH conditions (Fig. 4C, D, and 5 C, D). PaNuc-1 is stable for 24 h at temperatures between − 20 and 50 °C (> 85% activity) and displayed 13 ± 5% residual activity at 60 °C (Fig. 4C), whereas activity of PaNuc-2 is reduced to 67 ± 2% after incubation for 24 h at a temperature of 40 °C with < 25% residual activity being exhibited at 50 °C and no residual activity at 60 °C, respectively (Fig. 4D). The apparent kinetic parameters were determined with sheared dsDNA used as substrate (Table 2).

Substrate specificity of Pantoea agglomerans nucleases. Recombinant nucleases were incubated with different types of DNA and RNA. The degradation efficiency towards the expression plasmid pET24d::PaNuc-1 (Plasmid) was compared to unsheared dsDNA from calf thymus, to sheared dsDNA from salmon sperm, to ssDNA and to MS2 RNA, respectively

Effect of temperature on the activity and stability of recombinant PaNuc-1 and PaNuc-2. A, B Purified nucleases were incubated at different temperatures in 50 mM NaPO4 buffer pH 7.0 supplemented with 0.1% (w/v) bovine serum albumin (BSA). C, D The stability of the enzymes was measured after incubation for 24 h at different temperatures. Afterwards, activity was determined at 35 °C in 50 mM NaPO4 buffer pH 7.0, 0.1% (w/v) BSA as described in “Materials and Methods”. Error bars indicate standard deviations. All experiments were done in triplicate. Some deviation bars are that low that they are hidden behind the data point icons

Influence of pH on the activity and stability of PaNuc-1 and PaNuc-2. A, B To determine the optimal pH range for PaNuc-1 and PaNuc-2, enzyme assays were done in sodium acetate buffer (pH 4–6, open circles), sodium phosphate buffer (pH 5–7, open squares), and Tris-HCl buffer (pH 7–9, open diamonds). C, D Enzyme stabilities were examined after incubation for 60 min at different pH in sodium acetate buffer (pH 4–6, open circles), sodium phosphate buffer (pH 5–7, open squares), Tris-HCl buffer (pH 7–9, open diamonds), and glycine-NaOH buffer (pH 9–11, open triangles). Afterwards, activity was determined at 35 °C in 50 mM NaPO4 buffer pH 7.0, 0.1% (w/v) BSA as described in “Materials and Methods”. Error bars indicate standard deviations. All experiments were done in triplicate
Influence of mono- and divalent cations
Activity of recombinant nucleases is > 90% in the presence of EDTA up to concentrations of 20 mM in case of PaNuc-1 and up to 4 mM EDTA in case of PaNuc-2, respectively (Fig. 6). It is well accepted that salt concentrations in the reaction mixture influence the catalytic activity of metal-ion-dependent non-specific nucleases, while only little information is available for EDTA-tolerant nucleic acid degrading enzymes. Inhibitor studies using divalent metal ions at concentrations up to 10 mM revealed that PaNuc-1 is not impaired by MgCl2 (Fig. 7A), but a decrease in activity is detected in the presence of MnCl2 (Fig. 7B) and CaCl2 (Fig. 7C) at a concentration of 10 mM and at 2 mM of CoCl2 (Fig. 7D). In contrast to these results, PaNuc-2 is negatively influenced by 2 mM MgCl2 (Fig. 7A) and 0.1 mM of MnCl2 (Fig. 7B), respectively, and completely inactive in the presence of CaCl2 (Fig. 7C) and CoCl2 (Fig. 7D) at concentrations of 10 mM. The influence of monovalent ions K+ and Na+ was tested at concentrations between 50 and 1000 mM. A concentration of 250 mM KCl and NaCl partly inhibits PaNuc-1, while PaNuc-2 is impaired at a concentration of 100 mM KCl and 250 mM NaCl, respectively (Fig. 7E, F).

EDTA tolerance tests. A Control experiments: sheared salmon sperm DNA was incubated with Benzonase® Nuclease (Merck, Darmstadt, Germany) as positive control (+) and without nuclease (−). M – Marker. PaNuc-1 (B) and PaNuc-2 (C) were pre-incubated without (0 mM) or in the presence of different concentrations of EDTA (2–50 mM). Afterwards, sheared salmon sperm DNA was added to the reaction mixture to start activity assays. Qualitative assays are depicted on agarose gels, while quantitative measurements are indicated by line graphs. Error bars indicate standard deviations. All experiments were done in triplicate

Effect of monovalent and divalent metal ions on the catalytic performance of recombinant nucleases. Divalent metal ions MgCl2 (A), MnCl2 (B), CaCl2 (C), and CoCl2 (D) were tested in a concentration range between 0.1 and 10 mM, while KCl (E) and NaCl (F) were supplemented to the reaction buffer in a final concentration of 50 to 1000 mM
Influence of various chemical components on the activity of nucleases from Pantoea agglomerans
Both nucleases, PaNuc-1 and PaNuc-2, were neither impaired by the presence of serine modifiers nor by reducing agents, while organic solvents at concentrations of 50% (v/v) reduced the catalytic activities of both enzymes below 40% (Table 3). The addition of 1 M of the chaotrope guanidine-HCl completely inactivated nucleases, while 1 M urea reduced enzymatic activities to 87% ± 9% and 93% ± 3%, respectively. The influence of non-ionic surfactants, such as Tween 20 or Triton X-100, on nuclease activities, was not tested due to assay limitations resulting in solubilized nucleic acids that could not have been precipitated. The utilization of the detergent SDS at a concentration of 0.25% (w/v) inactivated both enzymes (Fig. S2).
Discussion
Nucleases were described to be relevant for versatile cellular functions including nutrition, recombination, and repair mechanisms or DNA replication (Rangarajan and Shankar 2001). Microorganisms that secrete extracellular nucleases can even use nucleic acids as the sole source of carbon. Evolution of versatile secreted homologs might have taken place in the family Enterobacteriaceae to respond to different environmental conditions. Two genes encoding functional non-specific nucleases were discovered in the genome of the plant pathogen competitor bacterium P. agglomerans, which might have been the result of an ancient gene duplication event. A widely accepted hypothesis indicates that ancestral proteins were promiscuous allowing specific refinement of functionalities in the course of evolution (Pandya et al. 2014). Both enzymes from P. agglomerans were active on all nucleic acids that were tested in this study, including ssDNA, dsDNA, RNA, and circular plasmids. At the same time, they displayed immense differences in their catalytic properties with regard to temperature activity ranges or sensitivity towards metal ions, detergents, or organic solvents.
Different isolates of P. agglomerans have been described to tolerate growth temperatures up to 42 °C, at which both extracellular NSN display activities of > 70%, indicating nearly optimal conditions, which might imply that these enzymes are important for nutrient supply in natural environments (Abbas et al. 2017; Costa et al. 2002). Nuclease-mediated hydrolysis of phosphodiester bonds in DNA is often strictly dependent on the presence of a divalent metal ion that is essential as a cofactor to catalyze the hydrolytic reaction (Yang 2011). Nowadays, only a few nucleases were described to be metal-ion-independent including GBSV1-NSN from a thermophilic bacteriophage and members of the PLD-like family of nucleases (Bao et al. 2008; Grazulis et al. 2005; Song and Zhang 2008; Stuckey and Dixon 1999). PLD-like nuclease Nuc from S. enterica subsp. enterica serovar Typhimurium is not impaired in the presence of metal ion chelators and not influenced by divalent metal ions. A concentration of 4 mM of EDTA did not affect PaNuc-1 at all (99% residual activity). A concentration of 10 mM EDTA inhibited the non-specific nuclease activity of the cleavage domain of another restriction endonuclease (BmrI) from Bacillus megaterium, while the residual activities of PaNuc-1 and PaNuc-2 are only reduced by 9% and 11%, respectively, compared to the sample without metal ion chelator (Bao et al. 2008). However, EDTA-tolerance is not a typical feature of enzymes from P. agglomerans. It was demonstrated that a gallic acid decarboxylase is inhibited by 72% in the presence of EDTA at a concentration of 1 mM (Zeida et al. 1998). In addition, PaNuc-2 is sensitive towards 10 mM of Mg2+ and Mn2+, while PaNuc-1 is slightly inhibited when the reaction buffer is supplemented with Ca2+ and Co2+. Moreover, both nucleases from P. agglomerans, although not derived from a halophilic bacterial source, display a certain grade of salt tolerance. Both enzymes exhibited catalytic activity in the presence of NaCl and KCl at concentrations up to 100–250 mM, which is comparable to the non-specific, metal-ion dependent endonuclease DNase A from Fibrobacter succinogenes (MacLellan and Forsberg 2001). However, both recombinant enzymes were sensitive to the presence of detergents. Extracellular nuclease from Pseudomonas BAL31 is still active in the presence of 5% (w/v) SDS as long as the reaction mixture is supplemented with the metal ions Ca2+ and Mg2+, whereas both nucleases from P. agglomerans were dramatically impaired in the presence of less than 0.25% (w/v) SDS (Gray Jr. et al. 1975).
In good agreement to the bacterial Nuc from S. enterica subsp. enterica serovar Typhimurium as well as the DNases from the E. coli bacterial toxin colicin E9 and the strict anaerobe F. succinogenes that were described to be active as monomers, non-specific nucleases from P. agglomerans also exhibited a monomeric form (MacLellan and Forsberg 2001; Pommer et al. 1998; Zhao et al. 1997). Moreover, the activity of the nuclease from F. succinogenes was strictly dependent on the formation of an intramolecular disulfide bond, while the presence or absence of a disulfide bond in the nuclease PaNuc-2 did not influence the catalytic performance of the enzyme, because the addition of reducing agents (up to 10 mM of dithiothreitol (DTT) or β-mercaptoethanol) had hardly any inhibitory effect (MacLellan and Forsberg 2001).
High-level production of recombinant proteins in E. coli often lead to the formation of intermolecular hydrophobic interactions that result in the formation of inclusion bodies (IBs). In such cases, isolated IBs are usually washed with detergents and solubilized in chaotropic agents. Afterwards, proteins are refolded at mild conditions and purified using chromatographic strategies (Singh et al. 2015). PaNuc-2 exhibits a theoretical pI of 6.9 and was successfully refolded in a buffer with an alkaline pH of 8.5, which is in good agreement with literature data stating that refolding efficiency increased with the difference between the pI of the protein and the average pH of the refolding buffer (MacLellan and Forsberg 2001). The PLD-domain of the homologous protein BNC1 from the EDTA-degrading bacterium Chelativorans multitrophicus was also not soluble in our hands when expressed in E. coli using the pET24d-expression vector (data not shown, (Doronina et al. 2010)).
Phylogenetic analyses led to the hypothesis that the two members of the NSN family are probably the result of an ancient gene duplication event and biochemical characterization of the recombinant enzymes indicated that the two proteins underwent functional diversification in the course of evolution. It has been shown that the screening for gene duplication events on the taxonomic family level or above, is a promising strategy for the identification of specific enzymes exhibiting versatile biochemical properties (Elleuche et al. 2013; Espinosa-Cantu et al. 2015; Pandya et al. 2014).
Commercially available NSN were shown to be of high value for the degradation of nucleic acids in protein purification downstream processes (Rangarajan and Shankar 2001). Such enzymes can also be applied in cell-sorting systems. The degradation of nucleic acids derived from lysed cells reduces the viscosity of buffer solutions in microfabricated channel systems and thereby prevent clogging effects (Miltenyi et al. 2018). Due to their catalytic properties, these enzymes might be demanded for potential biotechnological applications, when EDTA-containing buffers are applied at elevated temperatures and broad pH ranges to ensure nucleic acid–free process setups.
References
Abbas Z, Authman S, Al-Ezee A (2017) Temperature effects on growth of the biocontrol agent Pantoea agglomerans (an oval isolate from Iraqi soils). J Adv Lab Res Biol 8:85–88
Bao Y, Higgins L, Zhang P, Chan SH, Laget S, Sweeney S, Lunnen K, Xu SY (2008) Expression and purification of BmrI restriction endonuclease and its N-terminal cleavage domain variants. Protein Expr Purif 58:42–52
Costa E, Usall J, Teixido N, Delgado J, Vinas I (2002) Water activity, temperature, and pH effects on growth of the biocontrol agent Pantoea agglomerans CPA-2. Can J Microbiol 48:1082–1088
Cruz AT, Cazacu AC, Allen CH (2007) Pantoea agglomerans, a plant pathogen causing human disease. J Clin Microbiol 45:1989–1992
Doronina NV, Kaparullina EN, Trotsenko YA, Nortemann B, Bucheli-Witschel M, Weilenmann HU, Egli T (2010) Chelativorans multitrophicus gen. nov., sp. nov. and Chelativorans oligotrophicus sp. nov., aerobic EDTA-degrading bacteria. Int J Syst Evol Microbiol 60:1044–1051
Elleuche S, Klippel B, von der Heyde A, Antranikian G (2013) Comparative analysis of two members of the metal ion-containing group III-alcohol dehydrogenases from Dickeya zeae. Biotechnol Lett 35:725–733
Espinosa-Cantu A, Ascencio D, Barona-Gomez F, DeLuna A (2015) Gene duplication and the evolution of moonlighting proteins. Front Genet 6:227
Gray HB Jr, Ostrander DA, Hodnett JL, Legerski RJ, Robberson DL (1975) Extracellular nucleases of Pseudomonas BAL 31. I. Characterization of single strand-specific deoxyriboendonuclease and double-strand deoxyriboexonuclease activities. Nucleic Acids Res 2:1459–1492
Grazulis S, Manakova E, Roessle M, Bochtler M, Tamulaitiene G, Huber R, Siksnys V (2005) Structure of the metal-independent restriction enzyme BfiI reveals fusion of a specific DNA-binding domain with a nonspecific nuclease. Proc Natl Acad Sci U S A 102:15797–15802
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Leiros I, Secundo F, Zambonelli C, Servi S, Hough E (2000) The first crystal structure of a phospholipase D. Structure 8:655–667
Li L, Rohrmann GF (2000) Characterization of a baculovirus alkaline nuclease. J Virol 74:6401–6407
MacLellan SR, Forsberg CW (2001) Properties of the major non-specific endonuclease from the strict anaerobe Fibrobacter succinogenes and evidence for disulfide bond formation in vivo. Microbiology 147:315–323
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226
Miltenyi S, Hübel T, Nölle V (2018) Process for sorting cells by microfabricated components using a nuclease. USA Patent US 10,018,541 B2, Jul. 10, 2018
Nielsen H (2017) Predicting secretory proteins with SignalP. Methods Mol Biol 1611:59–73
Pandya C, Farelli JD, Dunaway-Mariano D, Allen KN (2014) Enzyme promiscuity: engine of evolutionary innovation. J Biol Chem 289:30229–30236
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Pommer AJ, Wallis R, Moore GR, James R, Kleanthous C (1998) Enzymological characterization of the nuclease domain from the bacterial toxin colicin E9 from Escherichia coli. Biochem J 334(Pt 2):387–392
Ponting CP, Kerr ID (1996) A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: identification of duplicated repeats and potential active site residues. Protein Sci 5:914–922
Rangarajan ES, Shankar V (2001) Sugar non-specific endonucleases. FEMS Microbiol Rev 25:583–613
Rudolph AE, Stuckey JA, Zhao Y, Matthews HR, Patton WA, Moss J, Dixon JE (1999) Expression, characterization, and mutagenesis of the Yersinia pestis murine toxin, a phospholipase D superfamily member. J Biol Chem 274:11824–11831
Schmitz S, Nölle V, Elleuche S (2019) A non-specific nucleolytic enzyme and its application potential in EDTA-containing buffer solutions. Biotechnol Lett 41:129–136
Singh A, Upadhyay V, Upadhyay AK, Singh SM, Panda AK (2015) Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb Cell Factories 14:41
Song Q, Zhang X (2008) Characterization of a novel non-specific nuclease from thermophilic bacteriophage GBSV1. BMC Biotechnol 8:43
Stuckey JA, Dixon JE (1999) Crystal structure of a phospholipase D family member. Nat Struct Biol 6:278–284
Yang W (2011) Nucleases: diversity of structure, function and mechanism. Q Rev Biophys 44:1–93
Zeida M, Wieser M, Yoshida T, Sugio T, Nagasawa T (1998) Purification and characterization of gallic acid decarboxylase from Pantoea agglomerans T71. Appl Environ Microbiol 64:4743–4747
Zhao Y, Stuckey JA, Lohse DL, Dixon JE (1997) Expression, characterization, and crystallization of a member of the novel phospholipase D family of phosphodiesterases. Protein Sci 6:2655–2658
Acknowledgements
We thank Stefan Edelburg for the help with the VICTOR™ X4 Multilabel Plate Reader.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 171 kb)
Rights and permissions
About this article

Cite this article
Schmitz, S., Börner, P., Nölle, V. et al. Comparative analysis of two non-specific nucleases of the phospholipase D family from the plant pathogen competitor bacterium Pantoea agglomerans. Appl Microbiol Biotechnol 103, 2635–2648 (2019). https://doi.org/10.1007/s00253-019-09644-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09644-y