
Abstract
Non-specific nuclease (NSN) can be applied in industrial downstream processing to remove nucleic acids from crude protein extracts or in cell-sorting systems to degrade nucleic acids derived from lysed cells. PsNuc from the ice-nucleating bacterium Pseudomonas syringae has the ability to decompose double- and single-stranded DNA in linear or circular form and RNA. It is not affected by the presence of metal-ion chelators such as EDTA and tolerates several protease inhibitors and reducing agents. A multiple sequence alignment of PsNuc with closely related enzymes (97–99% identity on the protein level) within the family Pseudomonaceae revealed the presence of only six amino acid residues that are variable in putative NSN from different members of the genus Pseudomonas. Single amino acid variants were produced in recombinant form in Escherichia coli, purified, and characterized. They showed similar activity compared to PsNuc, but a single variant even displayed an improved performance with an activity of > 20,000 U/mg at 35 °C, while amino acid residues S148 and V161 were found to be essential for enzymatic functionality. These results suggest that homologous nucleases from Pseudomonaceae display high activity levels in a metal-ion-independent manner and are therefore of interest for applications in biotechnology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enzymes that are capable of digesting nucleic acids can be assigned to the group of nucleases. Such nucleolytic enzymes are ubiquitously distributed and were characterized from prokaryotes, eukaryotes, and viruses [14, 17, 20]. The group of nucleic acid-hydrolyzing enzymes is assigned to EC 3.1.11.x + 3.1.31.x and includes exo- and endoribonucleases, site-specific and non-specific hydrolases, and enzymes that produce 5′- or 3′-phosphomonoesters. The majority of nucleases share a common property: they belong to the group of metalloenzymes, which implies that they act in a metal-ion-dependent manner [34]. However, isolated members of some groups of metal-ion-independent nucleases have been identified and characterized. Site-specific restriction enzymes include the restriction DNA glycosylase R.PabI from the hyperthermophilic archeon Pyrococcus abyssi or the restriction endonuclease LlaKI from Lactococcus lactis KLDS4 [2, 31]. Besides several site-specific restriction endonucleases, only a few non-specific nucleases (NSNs) have been reported to act in a metal-ion-independent manner, including an enzyme encoded by the gene wsv191 in the genome of the white spot syndrome virus or the protein GBSV1-NSN from a thermophilic bacteriophage [13, 28].
In addition, there are diverse proteins in the phospholipase D (PLD, EC 3.1.4.4) superfamily that exhibit versatile functionalities including genuine PLD-enzymes, cardiolipin synthases, helicase-like proteins, a murine toxin, viral proteins of unknown function, and site-specific and non-specific nucleases [6, 24, 35]. The site-specific restriction endonucleases BfiI from Bacillus firmus and BmrI from Bacillus megaterium were biochemically and/or structurally characterized and are composed of a typical PLD-motif and a DNA-binding domain [1, 9]. The prototype of a NSN of the PLD-superfamily is the enzyme Nuc from Salmonella enterica subsp. enterica serovar Typhimurium [29, 35]. Primary sequence analysis and identification of the conserved HxK(x)4D(x)6GSxN motif clearly assigns Nuc to the PLD-superfamily. Such enzymes mainly catalyze the hydrolysis of phosphatidylcholine to give phosphatidic acid and choline. PLD-enzymes from mammals contain two copies of the conserved motif arranged in a tandem orientation on a single polypeptide, while bacterial endonucleases and a helicase-like protein from Escherichia coli contain a single motif, which usually dimerizes [29].
NSNs are of great biotechnological interest. These enzymes can be used to type human pathogenic bacterial clades, they are routinely implemented in industrial downstream processing to remove nucleic acids from crude protein extracts. Furthermore, NSN can also be applied in cell-sorting systems for the degradation of nucleic acids derived from lysed cells to prevent clogging in microfabricated channels [18, 22, 25]. Due to the fact that molecular biological and biotechnological applications often take place at moderate or even at lowered temperatures, the aim of this study was to identify a NSN that is active at moderate temperatures. Recently, the first endonuclease I from an extremely psychrophilic microorganism has been identified and characterized in detail [15]. Pseudomonas syringae thrives in cold to moderate temperature habitats and is one of the most effective natural causatives of ice nucleation. Moreover, most isolates of P. syringae are plant pathogens, some are not pathogenic, but exist as commensals in plants. This bacterium exhibits a tolerance to ultraviolet light and some strains are even polyextremophilic. They combine extreme conditions including a xerotolerant lifestyle (adapted to dry environments) [33].
In this study, a metal-ion-independent nuclease of the PLD-superfamily has been identified in the genomes of different strains and variations of P. syringae and related Pseudomonads. A multiple sequence alignment revealed that these proteins are highly conserved despite a very diverse putative secretion peptide sequence. Seven protein variants that only differ in single amino acid residue substitutions were produced in recombinant form in E. coli, purified, and characterized. Several variants display high catalytic activity rates that are in a range of commercial nucleases, such as bacterial Benzonase™ Nuclease or bovine DNaseI. In contrast to these metal-ion-dependent nucleases, PsNuc tolerates concentrations of EDTA up to 50 mM and shows potential to be applied in industrial processes at moderate temperature conditions.
Materials and Methods
Strains and Culture Conditions
Escherichia coli strain NEB® 5-alpha (New England Biolabs, Frankfurt/Main, Germany) was used for plasmid maintenance and propagation and strain Veggie BL21 (DE3) (New England Biolabs, Frankfurt/Main, Germany) for protein production.
Computational Sequence Analysis
BLASTP searches were done to identify putative non-specific nucleases from cold and EDTA-tolerating bacterial species. Sequences of metal-ion-independent nucleases including bacterial Nuc from Salmonella enterica subsp. enterica serovar Typhimurium (PDB: 1BYS_A), viral GBSV1-NSN (ABK63794.1), and viral WSSV-NSN (AAL33195.1) were used as queries. Protein sequence data of a putative nuclease (WP_050543862.1) from the gram-negative, ice-nucleating enterobacterium Pseudomonas syringae were obtained from NCBI Entrez (http://www.ncbi.nlm.nih.gov/entrez). The prediction of signal peptides was carried out using SignalP 4.1 server [19]. Prediction of conserved domains was done using the tool CDS (Conserved domain search, https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Secondary structure predictions were done with the PSIPRED server and homology models were done with SWISS-MODEL using Nuc as a template [3, 11]. Visualization of protein structures and preparation of illustrations was accomplished with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
An evolutionary-based strategy was chosen to investigate naturally occurring variations of PsNuc. BLASTP and tBLASTN database searches were performed using PsNuc without its signal secretion peptide as query. Multiple sequence alignments were produced with the ClustalX 2.1 program [12]. Constructs containing singular point mutations were generated based on following conditions (i) a certain amino acid residue substitution occurred in more than a single annotated sequence, (ii) the annotation could be clearly assigned to the bacterial genus Pseudomonas (annotations derived from environmental sample sequencing projects were omitted), and (iii) amino acids were not replaced by functionally related residues. The variant PsNuc_G116A is the sole exception of these rules, because the replacement of similar amino acids at position 116 is in close proximity to the catalytic tunnel and might be important for maintenance of enzymatic function (data not shown).
Cloning of the Gene Encoding PsNuc
The gene encoding PsNuc and gene variants with single amino acid substitutions were codon-optimized and synthesized by ATUM (Newark, California, USA). Synthetic genes were digested with NcoI/AatII to enable ligation into pET24d(+) (Merck, Darmstadt, Germany) equipped with a 2xHIS sequence for cytosolic protein production to give plasmid pET24d-2HIS-PsNuc and variants. Sequence verification was done by GATC Biotech AG (Konstanz, Germany). The cloning strategy and construct design has been described in detail in our previous publications [26, 27].
Recombinant Expression and Purification
Escherichia coli expression strain Veggie BL21 (DE3) harboring an expression plasmid was grown in shaking flasks at 37 °C. Two strategies were tested and compared: (i) Gene expression was induced with a final concentration of 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) after an OD600 = 0.5–0.7 was reached. Cells were harvested 4 h after induction, or (ii) cells were grown without induction and harvested after 16 h of incubation. Cell lysis was achieved by sonication in 20 mM NaH2PO4, pH 8.0. Lysates were loaded onto a column equipped with Ni-Sepharose (GE Healthcare, Munich, Germany) for affinity chromatography. Prior, the column was equilibrated with 50 mM NaH2PO4, 50 mM NaCl, 5 mM imidazole, pH 7.0. After loading, the column was washed subsequently with 50 mM NaH2PO4, 50 mM NaCl, 50 mM imidazole, pH 7.0, and 50 mM NaH2PO4, 50 mM NaCl, 150 mM imidazole, pH 7.0, prior to protein elution using 50 mM NaH2PO4, 500 mM NaCl, and 250 mM imidazole, pH 7.0. Fractions containing the target protein were pooled and diluted with equilibration buffer 25 mM NaH2PO4, pH 6.0, until a conductivity of < 9 mS/cm was reached. Subsequently, the protein was purified with cation-exchange chromatography (SP Sepharose® Fast Flow, GE Healthcare, Munich, Germany). After washing (25 mM NaH2PO4, 300 mM NaCl, pH 6.0), the target protein was eluted with 25 mM NaH2PO4, 1 M NaCl, pH 6.0. Afterwards, the HIS-tag fusion protein was cleaved using a specific peptidase that was removed by a final affinity chromatography step. PD10 desalting columns (GE Healthcare, Munich, Germany) were used to replace the cation exchange elution buffer with storage buffer (25 mM NaH2PO4, 25 mM NaCl, pH 7.0). The Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Darmstadt, Germany) was used to measure protein concentrations. SDS-PAGE (ProGel Tris Glycine 4–20%, anamed Elektrophorese GmbH, Groß-Bierberau/Rodau, Germany) in an X Cell SureLock™ Mini-Cell Electrophoresis System (Thermo Fisher Scientific, Darmstadt, Germany) and Western blot using the nitrocellulose blotting membrane (GE Healthcare, Munich, Germany) in a semi-dry Western blotting system (Biometra, Göttingen, Germany) was used for visualization of recombinant proteins. The Immobilon™ Western HRP substrate (Merck, Darmstadt, Germany) in combination with an anti-HIS HRP antibody (Miltenyi Biotec B.V. & Co. KG) was applied to detect HIS-tagged nuclease variants.
Enzyme Activity Assays
Nucleolytic activity was detected in vivo by growing E. coli Veggie BL21 (DE3) producing recombinant nucleases on DNase-indicator plates supplemented with toluidine blue (Merck, Darmstadt, Germany). Nuclease activity was further evaluated by agarose gel electrophoresis. Approximately, 100–200 ng of enzyme was applied to digest 5 µg of DNA. Substrate specificity was tested using circularized plasmid DNA, single-stranded RNA from bacteriophage MS2 (Sigma-Aldrich, St. Louis, USA), single-stranded DNA (ssDNA) from calf thymus (Sigma-Aldrich, St. Louis, USA), and double-stranded genomic DNA (dsDNA), namely UltraPure™ Salmon Sperm DNA Solution (Thermo Fisher Scientific, Darmstadt, Germany). Standard assays were done at 25 °C in 50 mM NaH2PO4, pH 7.0. The buffer solution was supplemented with different metal ions or EDTA, respectively, to qualitatively investigate the influence of such chemicals.
In addition, quantitative measurements were done spectrophotometrically by calculating the degradation rate of DNA using the VICTOR™ X4 Multilabel Plate Reader (PerkinElmer, Rodgau, Germany) in combination with Corning® 96-well UV-transparent plates (Merck, Darmstadt, Germany) in a standardized assay. Activity of PsNuc over time revealed a typical initial lag phase, which has been interpreted in a way that the first products from DNA digestion do not give rise to hyperchromicity [21]. Therefore, the assay in the VICTOR™ X4 Multilabel Plate Reader took place at 25 °C in a reaction volume of 100 µl including 10 ng of purified enzyme, which was incubated for 10 min in reaction buffer (50 mM NaH2PO4, pH 7.0) supplemented with 0.5 µg of sheared dsDNA to perform endpoint measurements. Due to the instability of PsNuc at temperatures above 25 °C, no standard assays were done at higher temperatures except for the determination of the temperature optimum. Undigested DNA was precipitated by using a 1:1 volume of 4% (v/v) perchloric acid. Samples were incubated on ice for 45 min prior to centrifugation at 4 °C, 10 min, 13,000 rpm to precipitate insoluble polymeric DNA fragments. Soluble fragmented oligonucleotides in the supernatant were measured at 260 nm.
Metal ions, chelators, solvents, and other potential inhibitors were preincubated with the enzyme in substrate-free samples on ice for 60 min. Afterward, activity assays were done under standard conditions. Reactions were started by the addition of sheared dsDNA. To calculate relative activities, the enzymatic performance of an individual control experiment at standard conditions without supplements was set to 100%.
To evaluate the influence of different temperatures and/or pH on nuclease activities, tests were done at temperatures between 4 and 60 °C at an optimal pH of 7 and between pH 5 and 9 at 25 °C, respectively. PsNuc was incubated for 60 min on ice in buffers at different pH (between pH 4 and 11) to investigate its stability, prior to activity assays at standard conditions. To determine the thermal stability of the NSN, enzyme samples were incubated for 10 min at different temperatures (between − 20 and 50 °C) prior to an incubation on ice for 10 min followed by a standard assay. Reactions were started by the addition of sheared dsDNA. Midpoint denaturation temperatures as means of inflection temperature values (Ti) were recorded using a Tycho™ NT.6 (NanoTemper Technologies GmbH, Munich, Germany). According to the instructions of the manufacturer, the inflection temperature indicates the transitions in the folding state of a protein that results from changes in the intrinsic fluorescence of aromatic residues when a thermal ramp is applied.
To determine the potential phospholipase activities of the nuclease PsNuc, phosphatidylcholine together with the Amplex® Red Phospholipase D Assay Kit (Thermo Fisher Scientific, Pinneberg, Germany) was used as substrate and the artificial substrate bis(4-nitrophenyl)phosphate (Merck, Darmstadt, Germany) was tested. In both cases, the phospholipase D from Streptomyces chromofuscus (Merck, Darmstadt, Germany) was used as a positive control. All assays were done as described by the manufacturers and performed in triplicate.
Kinetics
Enzyme kinetics were determined using sheared dsDNA as described earlier [16]. In brief, a mean molar mass per nucleotide of 330 g/mol was used to convert concentrations of sheared dsDNA given in µg/ml to calculate molar concentrations. One unit of nuclease activity is defined as ΔA260 min−1 per mg protein. Vmax and KM were obtained from Michaelis–Menten by non-linear regression using the curve-fitting tool of the add-in “Solver” in Microsoft Excel. Kcat-values were generated from Vmax by the assumption that the degradation of 0.05 mg of DNA resulted in a change in absorbance of 0.3 using the general formula Kcat = Vmax/[E0] [16]. All measurements were done in triplicate.
Results
PsNuc is a Homologue of the EDTA-Compatible NSN from Salmonella typhimurium
In order to identify a metal-ion-independent and cold-active nuclease, a sequence-based screening approach was applied using the enzyme Nuc (WP_000731968.1) from S. enterica subsp. enterica serovar Typhimurium (formally known as Salmonella typhimurium) as template. Nuc belongs to the superfamily of phospholipase D (PLD) proteins. This group of enzymes is well studied in mammals with Nuc being the first characterized isozyme from Bacteria. In contrast to mammalian relatives, which are represented by a polypeptide of about 1000 amino acid residues, Nuc is exclusively composed of 177 amino acid residues containing a single PLD domain and a preceding signal peptide sequence for protein secretion [29]. Homologous sequences of putative PLDs were identified within multiple genera of the order Enterobacterales dominated by Citrobacter, Escherichia, Enterobacter, Klebsiella, and Serratia exhibiting 60–99% identity on protein level. A conserved open reading frame was also identified within the order Pseudomonadales. There is only a limited number of putative nucleases belonging to the superfamily of phospholipase D from Pseudomonads, which are deposited in databases. These proteins mainly display sequence identities between 97 and 99% among each other, while no homologues with identities exceeding 66% to the homologues from Pseudomonads were found in other bacterial families (retrieved 2019-04-04). The closest homologue outside the order Pseudomonadales is an annotated endonuclease from Escherichia coli (KZI53816.1), which is 65.3% identical on the amino acid level. Owing to the domain prediction and its sequence identity to Nuc (57.2% sequence identity in 159 amino acids overlap), an annotated endonuclease from the ice-nucleating bacterium Pseudomonas syringae was designated PsNuc (WP_050543862.1) (Fig. 1a).

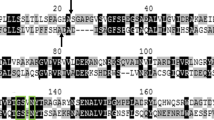
Structural properties of PsNuc. a ClustalX alignment of the non-specific nucleases PsNuc from Pseudomonas syringae and Nuc from Salmonella enterica subsp. enterica serovar Typhimurium. Identical and similar amino acids are highlighted by colons and dots. The predicted signal peptides are given in red letters and amino acid residues that are part of the conserved PLD-motif HxK(x)4D(x)6GSxN are boxed in magenta (catalytic region) or green (D129). Secondary structure elements that are either based on the solved structure of Nuc or predicted in PsNuc are indicated as arrows (β-strands) and barrels (α-helices). See Supplementary Fig. S1 for numbering of amino acid residues. b Schematic illustration of PsNuc. A hypothetical signal peptide (SP) composed of amino acid residues 1–30 is indicated. The domain PLDc-Nuc (amino acids 32–175 in PsNuc) is conserved in phospholipase D homologues. c Low-resolution homology model of PsNuc produced with SWISS-MODEL by using Nuc (PDB: 1BYS_A) as template. A hypothetical dimeric structure is illustrated with light and dark gray monomers. PyMOL was used to highlight conserved residues of the HxK(x)4D(x)6GSxN motif as sticks. Conserved residue D129 is boxed in green. Structure is rotated 90° along the X-axis to provide the bottom view of the catalytic region including conserved amino acid residues boxed in magenta (Color figure online)
The peptide sequence of PsNuc is composed of 183 amino acid residues with a predicted molecular weight of 19,972.74 Da and an isoelectric point of 9.4 (Table 1). A hypothetical signal peptide sequence was predicted to cover amino acid residues M1 to A30 (Fig. 1b, Supplementary Fig. S1). The molecular weight of the mature protein was predicted to be 16,826.93 Da with a calculated pI of 9.0. The characteristic domain of the PLD-superfamily (PLDc_Nuc) was assigned by the tool CDS to be located between amino acid residues E32 and Q175. These enzymes exhibit the conserved catalytic motif HxK(x)4D(x)6GSxN. An amino acid sequence alignment of PsNuc with Nuc from S. enterica subsp. enterica serovar Typhimurium in combination with secondary structure predictions and a homology model was used to identify the catalytic motif, which is composed of the amino acid residues H122, K124, G136, S137, and N139 in PsNuc (numbering starts with M1 of the predicted signal peptide), respectively. In addition, the conserved D129 is also part of the HxK(x)4D(x)6GSxN and has been described to form hydrogen bonds with the α1-β2-loop in Nuc [29] (Fig. 1a, c).
Production and Purification of the Recombinant NSN PsNuc in Escherichia coli
Escherichia coli strain Veggie BL21 (DE3) was transformed with an expression plasmid encoding an N-terminally HIS-tagged variant of NSN PsNuc. Enzymatic activity of PsNuc could be observed by DNase plate agar assays (Fig. 2a). Two expression strategies were tested: (1) induction of gene expression using IPTG resulted in cell death, while (2) non-induced cell growth for 16 h led to the production of recombinant protein. PsNuc was produced in soluble form and could be purified 23.1-fold to give a final activity of 6144 U/mg and resulted in a yield of 1.7% (Table S1). The unspecific cleavage of the first 19 amino acid residues was verified by a double protein band that occurred after affinity and ion exchange chromatography (Fig. 2b). Peptide mass fingerprinting led to the discovery of the exact position of the cleavage site (MGVNGSSHHHHHHSSGGGR/GSHHHHHH). Separation of the cleaved, tag-less enzyme revealed a signal with an apparent molecular weight of 17 kDa, which was confirmed by mass spectrometry analysis (16826.905, non-reduced conditions; 16827.005 Da, reduced conditions) (Supplementary Fig. S1).

Production and purification of the recombinant nuclease PsNuc a Toluidine blue DNase-indicator plates were used to grow E. coli Veggie BL21 (DE3) harboring plasmid pET24d-2HIS-PsNuc to produce PsNuc or E. coli Veggie BL21 (DE3) harboring plasmid pET24d as control. A bright pink zone indicates nucleolytic activity. Colonies were grown for 48 h. b Heterologous production and purification of Pseudomonas syringae PsNuc. E. coli Veggie BL21 (DE3) harboring plasmid pET24d-2HIS-PsNuc were grown for 24 h. Crude protein extracts from harvested cell cultures were produced by sonication and insoluble proteins in the pellet fraction (PE) were separated from soluble proteins in the supernatant (SN). A HIS-tagged variant of PsNuc was purified by a combination of affinity chromatography (Affinity) and ion-exchange chromatography (IEX) prior to proteolytic cleavage. Finally, pure enzyme (PsNuc) was separated from cleaved HIS-tags and pooled
Biochemical Properties of PsNuc
In vitro activity assays revealed that PsNuc is capable of hydrolyzing sheared and unsheared dsDNA, circular DNA, and linear ssDNA and ssRNA (Fig. 3a), but it is not active toward phospholipase D substrates such as phosphatidylcholine or bis(4-nitrophenyl)phosphate (Supplementary Fig. S2). The enzyme displays > 25% activity in a temperature range between 20 °C and 45 °C, including residual activity of 4% at 4 °C (Fig. 3b). The pH optimum is at pH 7.0, exhibiting > 50% activity between pH 6.0 to pH 8.0 (Fig. 3c). PsNuc is stable in a temperature range between − 20 °C and up to 20 °C (> 90% residual activity after incubation for 10 min) and in a pH range between pH 5.0 and pH 9.0 (Fig. 3b, c). Although, PsNuc is optimally active at temperatures above 40 °C, it becomes rapidly inactivated displaying 16% residual activity after incubation for 10 min at 40 °C (Fig. 3b). Prolonged incubation completely abolished the catalytic performance of this NSN. The inflection temperature was measured to be Ti = 54.4 °C (data not shown).

Substrate specificity, EDTA and salt stress, and influence of temperature and pH on the activity of PsNuc. a Substrate specificity of PsNuc. Different substrates were either incubated with (“+”) or without (“−”) recombinant PsNuc. Afterward, samples were loaded onto an agarose gel. Marker (M). Results indicate that PsNuc is capable of degrading sheared and unsheared genomic DNA, plasmid DNA, single-stranded DNA, and single-stranded RNA from the bacteriophage MS2. b Blue graph: Purified PsNuc was tested at optimal pH conditions (pH 7.0) in a temperature range between 4 and 60 °C using sheared dsDNA from salmon sperm as substrate. Black graph: For determination of the thermal stability of PsNuc, the recombinant protein was incubated for 10 min at different temperatures. Afterward, a standard assay (see “Methods”) was performed under optimal conditions (pH 7.0, 25 °C). c Blue graphs: Effect of the pH on PsNuc was tested by calculating relative activities in different buffers at a temperature of 25 °C. Black graphs: In order to determine the pH stability, PsNuc was incubated for 60 min on ice in different buffers. Subsequently, a standard assay (see “Methods”) was done. The following buffers were used: 50 mM Na-Acetate (filled circles, pH 4–6), 50 mM NaPO4 (filled squares, pH 5–8), 50 mM Tris/HCl (filled diamonds, pH 7–9), and 50 mM Glycine–NaOH (filled triangles, pH 9–11). Standard deviations indicate results from three independent measurements. Low deviation bars are hidden behind the data point icons. dPsNuc was preincubated on ice for 60 min without (0 mM) or in the presence of different concentrations of EDTA (2–50 mM). Afterward, sheared salmon sperm dsDNA was added to the reaction mixture to start activity assays. Qualitative assays are depicted on agarose gels, while quantitative measurements are indicated by line graphs. e, f Recombinant PsNuc was preincubated on ice for 60 min without (0 mM) or in the presence of different concentrations of NaCl (50–1000 mM, e) or KCl (50–1000 mM, f), respectively. The assay was started by the addition of sheared dsDNA from salmon sperm and incubation under standard conditions (25 °C, pH 7.0) (Color figure online)
Michaelis–Menten kinetics revealed a KM of 282 ± 60 µM and a Vmax of 6519 ± 1125 U/mg using sheared dsDNA as substrate (Table 2). The turnover rate kcat was 1756 ± 303 s−1 when incubated at 25 °C (Table 2) and reached values of > 5500 s−1 at 35 °C (Vmax > 20,000 U/mg). A decreased turnover rate of about 255 s−1 was recorded at 4 °C.
In order to test the influence of different ingredients, such as organic solvents, detergents, or protease inhibitors, on the catalytic activity, the recombinant enzyme was tested at optimal conditions in buffer supplemented with such components. PsNuc is not impaired by serine modifiers at reducing conditions tested here and in the presence of divalent metal ions, but displayed reduced activity profiles in the presence of organic solvents and chaotropic agents (Table 3). In addition, recombinant PsNuc tolerated chelating agents such as DOTA and EGTA up to concentrations of 50 mM with residual relative activities of > 85% (data not shown). In the presence of EDTA, the recombinant nuclease exhibited residual activities of > 90% at a concentration of 20 mM, while the activity was reduced to 67% ± 1% (Fig. 3d). The turnover rate of PsNuc at 25 °C in the presence of 4 mM EDTA was 1748 s−1. In contrast to these results, relative activity of PsNuc decreased with increasing concentrations of monovalent ions (Fig. 3e, f). PsNuc tolerated salt concentrations between 0 and 50 mM, but a concentration of 100 mM of NaCl or KCl resulted in a reduction of 12% and 7%, respectively, while concentrations of 1000 mM completely inactivated PsNuc.
Identification of Natural Variants of PsNuc within the Pseudomonads
Another BLASTP approach using PsNuc as query was applied to identify homologous proteins within the order Pseudomonadales. In total, 66 sequences that were annotated either as endonuclease or as PLD-family protein were identified. A multiple sequence alignment was generated and incomplete sequences (truncated sequences or sequences containing gaps) were deleted. Moreover, predicted signal peptide sequences were removed resulting in a multiple sequence alignment composed of 54 sequences, which were 97–100% identical (Supplementary Fig. S3). Amino acid variants that were only identified in a single sequence such as S64N, D94E, A117V, I126F, S137L, or W156R were neglected. Eight different single-nucleotide variations at seven different positions were present in at least two independent sequences including R53G, N95S, G116A, N148S, D157E, D157G, V161E, and T182P, respectively (Supplementary Fig. S4). The respective amino acid residues were mapped onto structural protein models based on the crystal structure of Nuc from S. enterica subsp. enterica serovar Typhimurium. Amino acids R53, N95, G116, and D157 are located on the protein surface of the PsNuc model structure, while N148 is inside the substrate tunnel, and V161 is close to the dimerization region (Fig. 4a, Supplementary Fig. S5). Genes encoding PsNuc and single amino acid variants were cloned and prepared for the expression in Escherichia coli Veggie BL21 (DE3). Variants D157E, but not D157G, as well as T182P at the penultimate position of the last flexible loop were omitted.

Evolutionary-based genetic variants of PsNuc. a Low-resolution homology model of PsNuc illustrating positions of evolutionary-generated amino acid variations. A light gray monomer indicates positions of amino acid residues (in magenta) in PsNuc, while the hypothetical conformation of natural amino acid variants is highlighted as blue sticks in the dark gray monomer. b Toluidine blue DNase-indicator plates were used to test nucleolytic activity of E. coli Veggie BL21 (DE3) transformed with variations of plasmid pET24d-2HIS-PsNuc to produce single-point mutation variants. A bright pink zone in the surrounding of the colonies indicates nucleolytic activity of variants R53G, N95S, G116A, and D157G, respectively, while N148S and V161E exhibited no activity. Colonies were grown for 24 h. c Western blot to analyze the production of HIS-tagged PsNuc-variants (2HIS-PsNuc: 29.0 kDa) in crude protein extracts of E. coli Veggie BL21 (DE3). d Silver-stained SDS-PAGE to analyze the purified, tag-less variants of PsNuc (PsNuc: 16.9 kDa). Variant V161E could not be purified to homogeneity (Color figure online)
Production, Purification, and Biochemical Properties of Natural Variants
Escherichia coli strain Veggie BL21 (DE3) was transformed with several constructs encoding nuclease variants. N148S and V161E exhibited normal growth, while R53G, N95S, G116A, and D157G displayed slowed growth accompanied by cell lysis, which was comparable to the production of wild-type PsNuc. In accordance with these results, neither variant N148S nor variant V161E displayed catalytic activity on DNase-indicator agar plates (Fig. 4b). Nevertheless, heterologous production of all constructs was visualized by Western blot analysis, resulting in signals of about 30 kDa (Fig. 4c), which is in agreement with the estimated molecular weight of the tagged products (29.0 kDa). All single-point mutation variants were produced and purified accordingly to the strategy described for PsNuc. However, variants N148S and V161E revealed only minor amounts of purified proteins with V161E not being purified to homogeneity (Fig. 4d).
The purified variants R53G, N95S, G116A, N148S, and D157G were tested in vitro for catalytic activity. As expected, in vitro assays using recombinant variant N148S confirmed inactivity, which is in agreement with the results obtained from DNase-indicator agar plate assays (Fig. 4b, Table 2). Kinetic analyses revealed that R53G, N95S, and G116A displayed equivalent activity levels compared with the wild-type enzyme PsNuc, while D157G exhibited a turnover rate and a maximal reaction velocity of 2290 ± 275 s−1 and Vmax of 8503 ± 1022 U/mg (Table 2). However, inflection temperature of D157G was decreased down to 51.1 °C. Tolerance toward EDTA did not differ between wild-type enzyme PsNuc and D157G. In accordance with these results, the turnover rate of D157G in the presence of 4 mM EDTA was estimated to be 2338 s−1.
Discussion
In modern biotechnology approaches, enzymes that withstand harsh conditions from various sources including hot and cold, acidic and alkalic, or salty environments are highly demanded because of their tremendous potential to be applied in industrial processes [7]. The chelator-tolerant enzyme PsNuc from P. syringae is a multifunctional and non-specific nuclease exhibiting catalytic activity toward DNA and RNA. Although P. syringae is not a psychrophilic bacterium per definition, most strains of P. syringae display an optimal growth temperature of 28 °C; however, several phytotoxins (virulence factors) of pathogenic strains were shown to be induced by cold-shocks—this bacterium is a known ice-nucleating bacterium and its nuclease displays some properties typical for psychrozymes including low thermostability and residual activity at low temperatures [10, 33]). In contrast to PsNuc, a nuclease from the psychrophilic microorganism Psychromonas ingrahamii 37 was active at cold temperatures (4 °C), but was highly thermostable (99 °C) at the same time [15]. Another reason to screen P. syringae and related species for EDTA-tolerant members of nucleases is the known resistance of several species within the Pseudomonads to EDTA [32]. Although mainly investigated with regard to its destabilizing effect on the outer membrane in EDTA-sensitive species, we hypothesized that extracellular enzymes of EDTA-resistant species might also tolerate the presence of EDTA. Therefore, single-point substitution variants that were conserved in at least two annotated genes in the databases were produced to evaluate the catalytic performance of the respective recombinant proteins.
Using this approach, two novel amino acid residues (N148 and V161) in NSN from P. syringae that are essential for catalytic activity were identified. Substitutions to S148 and E161 were found in two and three deposited sequences, respectively. It can be hypothesized that either (i) these variants are the result of sequencing errors or (ii) the enzyme became inactivated in the course of evolution. The negative effect cannot be suppressed by further amino acid substitutions, because the remaining peptide chain is completely identical to the prototype enzyme PsNuc. Site-directed mutagenesis was used before to characterize the conserved HxK(x)4D(x)6GSxN in Nuc from S. enterica subsp. enterica serovar Typhimurium [8]. It has been shown previously that the mutation of the essential histidine within the conserved HxK(x)4D(x)6GSxN motif to asparagine in Nuc from S. enterica subsp. enterica serovar Typhimurium revealed a reduction by a factor of 105 of the enzymatic catalytic activity [8]. This observation was also confirmed by mutating the respective amino acid residue of the human PLD1 [30]. Amino acid residue N148 in PsNuc is also located near the substrate tunnel and close to E150. Amino acid residue E150 is homologous to E144 in Nuc (numbering includes signal peptide sequences of both enzymes). This glutamate (E144) is in close proximity to the catalytic region and has been earlier described to be essential for catalytic activity in Nuc from S. enterica subsp. enterica serovar Typhimurium, because it forms a hydrogen bond with the active site residue S131 (S137 in PsNuc) [29]. A structural model of the homologue PsNuc indicates the presence of a putative hydrogen bond between N148 and E150, which cannot be formed when N148 is replaced by a serine (S148) residue (Fig. 5). In contrast to these results, it can be hypothesized that variant V161E is (i) inactive either because the formation of the functional active dimeric protein conformation is sterically hindered by the presence of the large glutamate in the dimerization domain or (ii) because of a potential disturbance of the hydrophobic interaction of V161 with other aliphatic residues. Amino acid residue E161 may also be in close proximity to I33 from the other monomer, which might impair the formation of the protein dimer (Fig. 5).

Hypothetical model of the inactive variants of PsNuc N148S and V161E. Middle: Low-resolution homology model of PsNuc with amino acid residues N148 and V161 highlighted in magenta and S137 and E150, which are essential for catalytic activity, indicated in red. Top: Replacement of the small amino acid residue V161 with large amino acid E161 might result in the disturbance of hydrophobic interactions and/or steric hindrance, impairing the formation of the dimeric structure. I33 has been included to demonstrate close proximity of amino acids involved in dimer formation. Possible rotamers are not considered in this illustration. Bottom: A close-up of the amino acid residue N148, which forms a hydrogen bond with E150 in this model structure. E150 forms another hydrogen bond with S137, which is part of the conserved motif HxK(x)4D(x)6GSxN. Mutagenesis to S148 most likely abolishes hydrogen bond formation in the model structure (Color figure online)
Intracellular recombinant production of PsNuc and related nucleases impairs the growth of the heterologous host. However, several bacterial nucleases have been shown to be inhibited in premature form to guarantee chromosome integrity. Such candidates just fold into their active form after being exported from the cytoplasm [4]. Our observation is in contrast to the recombinant production of full-length Nuc from S. enterica subsp. enterica serovar Typhimurium, whose presence did not impair the growth of E. coli BL21 (DE3) [8]. Based on the presence of the predicted signal peptides, these enzymes might be naturally secreted to circumvent toxic effects and to fulfill their specific tasks in nutrient sequestration in the surrounding environment. However, their cellular roles are often incompletely understood, but it has also been discussed that such nucleases are involved in recombination, repair, or replication processes [23]. Due to its high activity at low temperatures in combination with its broad substrate acceptance, the secreted NSN from the ice-nucleating bacterium P. syringae might also be important to sequester nutrients composed of nucleic acids from the environment for the regulation of cellular growth.
In accordance with their multiple cellular roles, nucleases exhibit diverse substrate preferences and activities. However, only three members of the bacterial PLD-superfamily were recently investigated with regard to bioprocess conditions in terms of buffers, pH, temperature, or inhibiting and activating compounds, but neither an isozyme from E. coli nor two candidates from Pantoea agglomerans display such high activity levels as PsNuc and are active at temperatures below 10 °C [26, 27]. It has been shown earlier that metal-ion-dependent nucleases are impaired by increasing concentrations of metal ions and a group of cyanobacterial non-specific nucleases are even produced in the presence of an inhibitor that interacts with the nuclease and interferes with its catalytic and toxic activity [17]. In contrast to such metal-ion-dependent nucleases, PsNuc and its variants are not influenced by the presence of divalent metal ions and tolerate EDTA and other chelators, such as EGTA, at concentrations up to 50 mM. However, PsNuc is impaired by increasing concentrations of monovalent metal ions, including NaCl and KCl.
The Yersinia pestis murine toxin PLD-superfamily enzyme did not even exhibit any activity toward DNA, but catalyzed the hydrolysis of phosphatidylcholine and was active toward artificial substrates containing a phosphodiester bond including bis(para-nitrophenol)phosphate (pNPP) [24]. Nuc from S. enterica subsp. enterica serovar Typhimurium was shown to be active toward both nucleic acids and the artificial substrate pNPP [35], while PsNuc did not catalyze the hydrolysis of pNPP. It is a well-established hypothesis that specific enzymatic functions are acquired in the course of evolution as a result of gene duplication events of multifunctional precursors followed by a distribution of those functions to the descendants. An ancient gene duplication event in the superfamily of bacterial PLD-endonucleases has recently been proposed based on the characterization of two genes encoding distantly related isozymes within the species P. agglomerans [26]. This idea is strengthened by the fact that the loss of a function through mutational events might be simpler than the acquisition of novel functionalities. Moreover, a fine-tuning process of an existing (but maybe rather weak) function might result in the development of “not-so-new” specificities of enzymes [5].
The enzyme PsNuc and some of the generated variants displayed a combination of distinct enzymatic properties with regard to EDTA-tolerance and catalytic activity at low temperatures that indicate their potential to be used in biotechnological applications such as production of crude protein extracts or cell-sorting systems. The enzyme variant D157G exhibited a reaction velocity above 7500 U/mg at 25 °C, which was at the peak of all variants that were compared in this study. This variant will now be used as a starting point to further increase catalytic activity by semi-rational protein design approaches.
Conclusion
To our knowledge, this is the first comprehensive characterization of a NSN within the genus Pseudomonas. The tested natural variants of the secreted NSN from the ice-nucleating bacterium P. syringae are highly active enzymes that tolerate harsh conditions as they are often predominating in industrial approaches.
Abbreviations
- AEBSF:
-
4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride
- dsDNA:
-
Double-stranded deoxyribonucleic acid
- DTT:
-
Dithiothreitol
- EDTA:
-
Ethylenediaminetetraacetic acid
- IPTG:
-
Isopropyl β-d-1-thiogalactopyranoside
- NSN:
-
Non-specific nuclease
- PMSF:
-
Phenylmethylsulfonyl fluoride
- RNA:
-
Ribonucleic acid
- ssDNA:
-
Single-stranded deoxyribonucleic acid
- SP:
-
Signal peptide
References
Bao, Y., Higgins, L., Zhang, P., Chan, S. H., Laget, S., Sweeney, S., et al. (2008). Expression and purification of BmrI restriction endonuclease and its N-terminal cleavage domain variants. Protein Expression and Purification,58, 42–52.
Belkebir, A., & Azeddoug, H. (2012). Characterization of LlaKI, a new metal ion-independent restriction endonuclease from Lactococcus lactis KLDS4. ISRN Biochemistry,2012, 287230.
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., et al. (2014). SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research,42, W252–W258.
Binnenkade, L., Kreienbaum, M., & Thormann, K. M. (2018). Characterization of ExeM, an extracellular nuclease of Shewanella oneidensis MR-1. Frontiers in Microbiology,9, 1761.
Conant, G. C., & Wolfe, K. H. (2008). Turning a hobby into a job: How duplicated genes find new functions. Nature Reviews Genetics,9, 938–950.
Damnjanovic, J., & Iwasaki, Y. (2013). Phospholipase D as a catalyst: Application in phospholipid synthesis, molecular structure and protein engineering. Journal of Bioscience and Bioengineering,116, 271–280.
Elleuche, S., Schröder, C., Sahm, K., & Antranikian, G. (2014). Extremozymes–biocatalysts with unique properties from extremophilic microorganisms. Current Opinion in Biotechnology,29, 116–123.
Gottlin, E. B., Rudolph, A. E., Zhao, Y., Matthews, H. R., & Dixon, J. E. (1998). Catalytic mechanism of the phospholipase D superfamily proceeds via a covalent phosphohistidine intermediate. Proc Natl Acad Sci USA,95, 9202–9207.
Grazulis, S., Manakova, E., Roessle, M., Bochtler, M., Tamulaitiene, G., Huber, R., et al. (2005). Structure of the metal-independent restriction enzyme BfiI reveals fusion of a specific DNA-binding domain with a nonspecific nuclease. Proc Natl Acad Sci USA,102, 15797–15802.
Horikoshi, K., Antranikian, G., Bull, A. T., Robb, F. T., & Stetter, K. O. (2011). Extremophiles handbook (1st ed.). New York: Springer.
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. Journal of Molecular Biology,292, 195–202.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics,23, 2947–2948.
Li, L., Lin, S., & Yanga, F. (2005). Functional identification of the non-specific nuclease from white spot syndrome virus. Virology,337, 399–406.
Li, L., & Rohrmann, G. F. (2000). Characterization of a baculovirus alkaline nuclease. Journal of Virology,74, 6401–6407.
Maciejewska, N., Walkusz, R., Olszewski, M., & Szymanska, A. (2019). New nuclease from extremely psychrophilic microorganism Psychromonas ingrahamii 37: Identification and Characterization. Molecular Biotechnology,61, 122–133.
MacLellan, S. R., & Forsberg, C. W. (2001). Properties of the major non-specific endonuclease from the strict anaerobe Fibrobacter succinogenes and evidence for disulfide bond formation in vivo. Microbiology,147, 315–323.
Meiss, G., Franke, I., Gimadutdinow, O., Urbanke, C., & Pingoud, A. (1998). Biochemical characterization of Anabaena sp. strain PCC 7120 non-specific nuclease NucA and its inhibitor NuiA. European Journal of Biochemistry,251, 924–934.
Miltenyi, S., Hübel, T., & Nölle, V. (2018). Process for sorting cells by microfabricated components using a nuclease. USA Patent US 10018541 B2, July 10, 2018.
Nielsen, H. (2017). Predicting secretory proteins with signalP. Methods in Molecular Biology,1611, 59–73.
Nilsen, I. W., Overbo, K., Jensen Havdalen, L., Elde, M., Gjellesvik, D. R., & Lanes, O. (2010). The enzyme and the cDNA sequence of a thermolabile and double-strand specific DNase from Northern shrimps (Pandalus borealis). PLoS ONE,5, e10295.
Pommer, A. J., Wallis, R., Moore, G. R., James, R., & Kleanthous, C. (1998). Enzymological characterization of the nuclease domain from the bacterial toxin colicin E9 from Escherichia coli. Biochemical Journal,334(Pt 2), 387–392.
Rangarajan, E. S., & Shankar, V. (2001). Sugar non-specific endonucleases. FEMS Microbiology Reviews,25, 583–613.
Rangarajan, S., & Shankar, V. (1999). Extracellular nuclease from Rhizopus stolonifer: purification and characteristics of single strand preferential—deoxyribonuclease activity. Biochimica et Biophysica Acta,1473, 293–304.
Rudolph, A. E., Stuckey, J. A., Zhao, Y., Matthews, H. R., Patton, W. A., Moss, J., et al. (1999). Expression, characterization, and mutagenesis of the Yersinia pestis murine toxin, a phospholipase D superfamily member. Journal of Biological Chemistry,274, 11824–11831.
Schaumburg, F., Pauly, M., Schubert, G., Shittu, A., Tong, S., Leendertz, F., et al. (2014). Characterization of a novel thermostable nuclease homolog (NucM) in a highly divergent Staphylococcus aureus clade. Journal of Clinical Microbiology,52, 4036–4038.
Schmitz, S., Börner, P., Nölle, V., & Elleuche, S. (2019). Comparative analysis of two non-specific nucleases of the phospholipase D family from the plant pathogen competitor bacterium Pantoea agglomerans. Applied Microbiology and Biotechnology,103, 2635–2648.
Schmitz, S., Nölle, V., & Elleuche, S. (2019). A non-specific nucleolytic enzyme and its application potential in EDTA-containing buffer solutions. Biotechnology Letters,41, 129–136.
Song, Q., & Zhang, X. (2008). Characterization of a novel non-specific nuclease from thermophilic bacteriophage GBSV1. BMC Biotechnology,8, 43.
Stuckey, J. A., & Dixon, J. E. (1999). Crystal structure of a phospholipase D family member. Natural Structural Biology,6, 278–284.
Sung, T. C., Roper, R. L., Zhang, Y., Rudge, S. A., Temel, R., Hammond, S. M., et al. (1997). Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO Journal,16, 4519–4530.
Wang, D., Miyazono, K. I., & Tanokura, M. (2016). Tetrameric structure of the restriction DNA glycosylase R.PabI in complex with nonspecific double-stranded DNA. Scientific Reports,6, 35197.
Wilkinson, S. G. (1967). The sensitivity of pseudomonads to ethylenediaminetetra-acetic acid. Journal of General Microbiology,47, 67–76.
Xin, X. F., Kvitko, B., & He, S. Y. (2018). Pseudomonas syringae: What it takes to be a pathogen. Nature Reviews Microbiology,16, 316–328.
Yang, W. (2011). Nucleases: Diversity of structure, function and mechanism. Quarterly Reviews of Biophysics,44, 1–93.
Zhao, Y., Stuckey, J. A., Lohse, D. L., & Dixon, J. E. (1997). Expression, characterization, and crystallization of a member of the novel phospholipase D family of phosphodiesterases. Protein Science,6, 2655–2658.
Acknowledgements
This work was funded by Miltenyi Biotec B.V. & Co. KG. The authors thank Jens Helmer (Miltenyi Biotec B.V. & Co. KG) for peptide mass fingerprinting, mass spectrometry analysis, and for the preparation of Supplementary Fig. S1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SS, MW, VN, and SE are employees of Miltenyi Biotec B.V. & Co. KG.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Schmitz, S., Wieczorek, M., Nölle, V. et al. Characterization of Single Amino Acid Variations in an EDTA-Tolerating Non-specific Nuclease from the Ice-Nucleating Bacterium Pseudomonas syringae. Mol Biotechnol 62, 67–78 (2020). https://doi.org/10.1007/s12033-019-00229-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00229-8