
Abstract
Nitrilases are of commercial interest in the selective synthesis of carboxylic acids from nitriles. Nitrilase induction was achieved here in three bacterial strains through the incorporation of a previously unrecognised and inexpensive nitrilase inducer, dimethylformamide (DMF), during cultivation of two Rhodococcus rhodochrous strains (ATCC BAA-870 and PPPPB BD-1780), as well as a closely related organism (Pimelobacter simplex PPPPB BD-1781). Benzonitrile, a known nitrilase inducer, was ineffective in these strains. Biocatalytic product profiling, enzyme inhibition studies and protein sequencing were performed to distinguish the nitrilase activity from that of sequential nitrile hydratase-amidase activity. The expressed enzyme, a 40-kDa protein with high sequence similarity to nitrilase protein Uniprot Q-03217, hydrolyzed 3-cyanopyridine to produce nicotinic acid exclusively in strains BD-1780 and BD-1781. These strains were capable of synthesising both the vitamin nicotinic acid as well as β-amino acids, a compound class of pharmaceutical interest. The induced nitrilase demonstrated high enantioselectivity (> 99%) in the hydrolysis of 3-amino-3-phenylpropanenitrile to the corresponding carboxylic acid.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrilase enzymes (EC 3.5.5.1) belong to a superfamily of enzymes that are found widely distributed in nature (Brenner 2002), and consist of proteins constituted of homo-dimeric building blocks with αββα-αββα sandwich folds that contain an active site displaying conserved cysteine, glutamic acid and lysine residues. C-site interactions between nitrilase homodimers generate an unusual spiral polymer structure that may only then be catalytically active (Thuku et al. 2009). Nitrilases catalyse the hydrolysis of aliphatic, aromatic as well as aryl-aliphatic nitriles, generating a carboxylic acid product with the concomitant release of ammonia (Fig. 1), and are therefore of commercial value as biocatalysts in synthetic chemistry (Gong et al. 2012; Martínková et al. 2017).

Schematic diagram showing nitrilase activity in comparison to the nitrile hydratase-amidase coupled reaction pathway
A parallel nitrile hydrolysing reaction involving sequential action of a nitrile hydratase and amidase may be present in the same organism (Fig. 1). Unlike nitrile hydratase (EC 4.2.1.84), which is often constitutively expressed (Cramp and Cowan 1999; Nagasawa et al. 1986; Yamada and Kobayashi 1996; Endo and Watanabe 1989) (Table S1), nitrilase usually requires induction (Robinson and Hook 1964; Harper 1977; Lévi-Schil et al. 1995) with only a handful of strains demonstrating constitutive nitrilase enzyme production (Zheng et al. 2008, 2011; Gradley et al. 1994; Stalker et al. 1988; Gong et al. 2012; Fang et al. 2015; Ahmed et al. 2011) (Table S2). A number of nitrile-containing nitrilase inducers have been discovered, including benzonitrile, which was found to be an effective nitrilase enzyme inducer in Alcaligenes faecalis MTCC 10757 (Nageshwar et al. 2011); acetonitrile in Stenotrophomonas maltophilia AC21 (Badoei-Dalfard et al. 2016); 2-cyanopyridine in Fusarium solani IMI96840 (Vejvoda et al. 2010); tetrachloroterephthalonitrile (He et al. 2014) and propionitrile (Prasad et al. 2007) in Rhodococci; as well as isovaleronitrile in Rhodococcus rhodochrous J1 (Kobayashi et al. 1989) and Nocardia globerula NHB-2 (Sharma et al. 2011) (Table S2). However, these nitrile-based inducers may be subject to hydrolysis by the enzyme they induce, thereby inactivating them, and hence other classes of inducers are of interest. ε-Caprolactam, although ineffectual in organisms such as Nocardia globerula NHB-2 (Sharma et al. 2011), has been shown to induce nitrilase expression in R. rhodochrous J1 (Nagasawa et al. 1990) and Fusarium proliferatum (Yusuf et al. 2013) AUF-2 .
Biocatalytic studies of nitrile hydrolysis in wild-type organisms are complicated by a number of factors that hinder unambiguous identification of the responsible enzyme system. Firstly, there are the two competing biocatalytic systems (nitrilase and nitrile hydratase-amidase) that can generate the same carboxylic acid product (Fig. 1). In many microorganisms, including Rhodococci, both of these enzyme systems may be located together on the genome (Martínková et al. 2009) and may also be constitutively expressed. Secondly, a nitrilase may be expressed but remain inactive if not oligomerised (Thuku et al. 2009). Thirdly, the enzymes are promiscuous: the formation of the amide intermediate, typically resulting from the action of a nitrile hydratase, may be the product of a nitrilase under certain conditions where the double hydrolysis reaction does not go to completion (Fernandes et al. 2006; Mateo et al. 2006); while a highly purified R. rhodochrous J1 amidase has been shown to slowly hydrolyse benzonitrile to benzoic acid (Kobayashi et al. 1998). Hence, a combination of chemical and molecular analyses is required to elucidate the responsible biocatalyst.
Herein, we demonstrate that dimethylformamide (DMF) is a cheap and effective nitrilase inducer in Rhodococcus rhodochrous and the closely related Pimelobacter simplex. Moreover, we definitively determined that nitrilase was responsible for nicotinic acid production using combined biocatalytic reaction profiling, inhibition studies and molecular analysis. This is the first time that DMF has been shown to induce nitrilase.
Materials and methods
Isolates
Three bacterial strains, namely Rhodococcus rhodochrous ATCC BAA-870, R. rhodochrous strain PPPPB BD-1780 as well as Pimelobacter simplex PPPPB BD-1781, were assessed in the current study. The two PPPPB strains are deposited at the Plant Pathogenic and Plant Protecting Bacteria Culture Collection (ARC, Pretoria, South Africa) while the ATCC strain is deposited at the American Type Culture Collection (Virginia, USA). BAA-870 was previously isolated from an industrial soil sample (Brady et al. 2004) and strains BD-1780 and BD-1781 were previously isolated from agricultural soil samples in the Gauteng province, South Africa (Rapheeha et al. 2017). All cells were cryopreserved in phosphate buffer (100 mM, pH 7.2) containing a 20% (v/v) final glycerol concentration.
Cell cultivation and induction
All three strains were inoculated into tryptone soy broth (TSB) media for 72 h at 28 °C to allow for biomass production (0.5% inoculum volume, agitation at 180 rpm). Aseptic cell harvesting was done by centrifugation (10,000×g, 15 min) prior to biomass transfer into minimal media (Nagasawa et al. 1990) (control) or induction minimal media containing either 5 mM benzonitrile (BZN) or 0.5% v/v DMF (99%, Merck). All induction flasks were incubated for up to 72 h at 28 °C prior to cell harvesting by centrifugation (10,000×g, 15 min) and subsequent storage of the pellet at − 20 °C.
Subsequent hydrolysis reactions were carried out in 1 mL with 100 mg wet weight cell paste (21 mg dry cell wt) in the presence of 4.16 mg (40 mmol/L) of 3-CP and agitation at 28 °C for 1 h.
Biocatalysis reactions
3-Cyanopyridine (3-CP) biocatalysis reactions involved reacting 200 mg wet weight cell paste (42 mg dry wt) in 4 mL of 0.1 M TRIS buffer, pH 9.0) cell preparation with 75.0 mg 3-CP in 1 mL methanol (reaction final volume 5 mL) with agitation at 28 °C for 15 min. Reaction quenching was by sample acidification with 1 M HCl.
Product recovery
To extract nicotinamide, the reaction mixture was basified using a 1 M sodium hydroxide solution to a pH of 10.0 and then extracted (3× sample volume of ethyl acetate) in a mini-separation funnel. The combined organic phase was dried over anhydrous magnesium sulphate prior to product concentration by rotary evaporation. Flash column chromatographic purification allowed for the separation of the nitrile and the amide product.
Nicotinic acid extraction was done using a 1 M hydrochloric acid solution to acidify the reaction mixture to a final pH of 3.5–4 to protonate the carboxylic acid, followed by extraction into an organic phase (3× sample volume of n-butanol). Product concentration was achieved by removal of n-butanol in vacuo.
Sample preparation for NMR analysis
Nicotinamide and nicotinic acid samples were solubilised in 200 μL of deuterated methanol prior to the addition of 400 μL of deuterated chloroform. Samples were analysed using a 400-MHz Varian Inova NMR spectrometer operating at 399.939 MHz (1H) and 100.574 MHz (13C) at 30 °C. Spectra were referenced against tetramethylsilane (TMS, δ 0.00 ppm) for 1H NMR, and against residual protonated methanol (δ 49.1 ppm) for 13C NMR, see Figs S1.1-8 for NMR spectra.
HPLC analysis method
Nicotinic acid, nicotinamide and 3-CP present in the reaction mixtures were quantified by HPLC using a Waters Agilent 1100 (USA) system equipped with a binary pump, auto sampler and column thermostat. Data processing was done using Chemstation chromatography management data software. Separation was achieved using a C18 reverse phase column (Waters Spherisorb 5 cm ODS2 column (250 mm × 4.6 mm)), temperature controlled at 30 °C. The volume injected onto the column was 10 μL. The mobile phase was a binary gradient prepared from aqueous KH2PO4 solution (10 mM, pH 2.8) against acetonitrile solution (HPLC grade). An initial ratio of 90:10 (% v/v) was run for 6.0 min. Then, the gradient was ramped to a 30:70 (% v/v) aqueous: organic gradient for 5 min. The flow rate used was 1.0 mL/min (Binfeng et al. 2011). Detection was at 230 nm.
Calibration curves were constructed for nicotinic acid (1–20 mM), nicotinamide (1–10 mM) and 3-CP (1–5 mM) with correlation coefficients (R2) of 0.990, 0.998 and 0.995, respectively. The retention times for nicotinic acid, nicotinamide and 3-CP were 3.541, 3.955 and 10.660 min, respectively.
Enantioselective biocatalytic resolution of β-aminonitriles
DMF-induced cells (100 mg wet wt) were re-suspended in a 0.1 M TRIS buffer solution (pH 9) prior to the addition of 10 mM of 3-amino-3-phenylpropanenitrile or 3-amino-3-(4-methoxyphenyl)propanenitrile. The reaction mixture (final volume 1 mL) was stirred at 28 °C with amide formation monitored by TLC. Biocatalytic reactions were terminated by acidification of the reaction (1 M HCl) prior to cell centrifugation (10,000×g, 5 min). The supernatant fraction was analysed by chiral HPLC.
Chiral HPLC was performed using a Hewlett Packard 1100 system consisting of a binary pump, autosampler, column thermostat compartment, diode array detector and Chemstation chromatograph data management software, version B.03.02. The analytical column used for separation was a Chiralpak AD-H, 250 mm × 4.6 mm, 5 μm (Chiral Technologies Europe, France), conditioned for 1 h. The mobile phase used was a mixture of n-hexane and isopropanol (both analytical grade) in the volumetric ratio of 90:10. The flow rate used was 1.00 mL/min; column temperature was set at 25 °C. Detection was at 210 nm for 30 min.
Mass spectrometric analysis
Ultra-performance liquid chromatography mass spectrometry
A Waters UPLC coupled in tandem to a Waters SYNAPT G1 HDMS mass spectrometer was used to generate accurate mass data. Optimisation of the chromatographic separation was done utilising a Waters HSS T3 C18 column (150 mm × 2.1 mm, 1.7 μm) with the column temperature controlled at 60 °C. A binary solvent mixture was used consisting of aqueous formic acid (10 mM) (eluent A) (natural pH of 2.3) and 10 mM formic acid in methanol (eluent B). The initial conditions were 98% eluent A at a flow rate of 0.4 mL/min which was maintained for 5 min, followed by a linear gradient to 5% eluent A at 6 min. The conditions were kept constant for 1 min and then re-set to the initial conditions. The run time was 10 min and the sample injection volume was 1 μL.
Time-of-flight mass spectrometric analysis (Tof-MS)
A SYNAPT G1 mass spectrometer was used in V-optics and operated in electrospray mode. Leucine enkephalin (50 pg/mL) was used as reference calibrant to obtain typical mass accuracies between 1 and 5 mDa. The mass spectrometer was operated in ESI positive and negative mode with a capillary voltage of 4.0 kV, the sampling cone at 10 V and the extraction cone at 3.0 V. The scan time was 0.2 s covering the 50 to 650 Da mass ranges. The source temperature was 120 °C and the desolvation temperature was set at 450 °C. Nitrogen gas was used as the nebulisation gas at a flow rate of 550 L/h and cone gas was added at 50 L/h. MassLynx 4.1 (SCN 872) software was used to control the hyphenated system and for data manipulation.
Enzyme inhibition
This was based on the inhibition of reactions converting 3-CP to nicotinic acid. Reactions were carried out using 100 mg of DMF-induced cells (wet wt) with nitrile degrading activity quantification performed using 20 mM 3-CP as substrate (final reaction volume 1 mL). All reactions were incubated at 28 °C for 60 min. Hydroxylamine (10 mM) (Prasad et al. 2009), ascorbic acid (10 mM) (Borsook et al. 1937) and nickel chloride (5 mM) were added to separate reactions to inhibit nitrile hydratase activity. Urea (5%) (Gregoriou and Brown 1979; Thalenfeld and Grossowicz 1976) and DEPA (7 mM) (Bauer et al. 1998; Brady et al. 2004) were included into reactions as aliphatic amidase inhibitors. Nitrilase competitive inhibitors benzylamine and benzaldehyde (10 mM) (Stevenson et al. 1992) were used to confirm the presence of nitrilase activity. Reactant concentrations were determined by HPLC. Inhibition was determined as the percentage decrease in the yield of nicotinic acid compared to the control reaction.
Protein isolation and SDS-PAGE analysis
Cell disruption
All control and induced cells were disrupted using a modified cell lysis method based on the protocol by Goncalves et al. (2006). Approximately 200 mg cell paste (wet wt) of the control, BZN and DMF-induced resting cells (72 h old induced cells) were re-suspended in 2 mL TRIS buffer (0.1 M, pH 8.8). Sodium dodecyl sulphate was added to the samples to a final concentration of 2.5% (v/v), prior to the addition of a protease inhibitor cocktail tablet into each sample [cOmplete ULTRA tablets, mini EDTA-free, Cat No.: 05892791001]. Glass beads (0.5 mm diameter, BioSpec Products, Cat No.: 11079105) were added into each tube to a final volume of 5 mL, with each subjected to 5 cycles of vortexing for 1 min followed by storage on ice for 1 min. Samples were subjected to vortexing and heating at 64 °C for 10 cycles of 1 min each. The cell lysate was centrifuged (13,000×g, 5 min) prior to storage of the samples at − 20 °C. Further characterisation of the cell lysate was then done by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE).
Protein sample denaturation
Protein samples were incubated at 70 °C for 10 min in the presence of a 1× reducing agent (Bolt ™ sample reducing buffer, Cat No.: 1772696) and a 1× loading sample buffer (Bolt ™ LDS sample loading buffer, Cat No.:1772823) prior to protein separation on a commercially prepared denaturing SDS-PAGE gel.
SDS-PAGE gel profiling
Denaturing SDS-PAGE gel electrophoresis was done using the method of Laemmli (1970). A BSA standard curve (100–800 ng/10 μL) was used for quantification purposes. All samples were loaded onto a precast 4–12% PAGE gel [Bolt ™ 4–12% Bis-TRIS Plus, Cat No.: NW04125, LTC scientific] with each sample undergoing denaturation in the presence of a reducing agent at 70 °C for 10 min prior to loading onto the gel, and running under constant voltage conditions (200 V) using a commercial morpholinoethanesulfonic acid buffer [Bolt™ MES buffer, Cat No.: 1771547]. Electrophoresis was performed at room temperature until the loading dye had migrated to the bottom of the gel. Gel staining was done using a Coomassie B-250 stain solution. Molecular weight determination was done using the Fermentas Pst1 molecular weight marker (Thermofischer, PageRuler Plus Prestained Protein ladder-Thermoscientific, Cat No.: 26634). The Biorad Chemi Doc™ imaging system with Image Lab 4.1 software was used to determine the intensity of the bands and quantified against a BSA standard curve. Bands were excised from each of the protein gels and submitted for in-gel digestion prior to LC-MSMS sequence analysis.
Sample preparation for peptide sequencing by LC-MSMS
In-gel digestions
SDS-PAGE excised gel slices were diced into ~ 1 mm2 fragments, with 200 μL of 50 mM NH4HCO3 in 50% methanol being added into each sample, and vortexed for 20 min before discarding the supernatant. This was repeated to allow for adequate gel de-staining prior to the addition of 100 μL of 75% acetonitrile. The resulting sample was then vortexed for 20 min with the supernatant being discarded and the gel pieces dried under vacuum. Protein plugs were stored at − 20 °C prior to sample denaturation and alkylation.
Alkylation
Approximately 25 μL of freshly prepared 10 mM dithiothreitol (DTT) prepared in 25 mM NH4HCO3 was added to each gel slice prior to vortexing. The protein sample was then incubated at 60 °C for 1 h to allow for sample denaturation and reduction before being cooled to room temperature. Approximately 500 μL of MS grade acrylonitrile was added to the samples, which were incubated at room temperature for a further 10 min with the supernatant being discarded. Approximately 25 μL of a 55 mM iodoacetamide solution prepared in 25 mM NH4HCO3 was added to the protein gel pieces before vortexing and incubation at room temperature for 20 min. The supernatant was discarded and the gel pieces were washed with 100 μL NH4HCO3 prepared in milli-Q water. The supernatant was once again discarded and the individual gel pieces dehydrated by the addition of 100 μL of a 50% acrylonitrile in 25 mM NH4HCO3 solution. Protein plugs were dried under vacuum and stored at − 20 °C.
Trypsin digestion
This was performed by incubating the protein plug with 1–5 ng/μL MS grade trypsin. Approximately 50 μL of 25 mM NH4HCO3 was added to each sample prior to incubation overnight at 37 °C to facilitate protein cleavage. Peptide extraction was carried out in protein Lo-Bind tubes (Eppendorf, Cat No.: 0030108035). Approximately 30 μL of 50% acrylonitrile/5% formic acid in milli-Q deionised water was added to the protein plugs with the samples being vortexed for 30 min at room temperature. All samples were centrifuged and then sonicated for 5 min prior to collection of the digest solution and vacuum drying before analysing by LCMS-MS.
LCMS-MS analysis
MS-derived proteomics analysis was done using the CompOmics PeptideShaker programme (Vaudel et al. 2015). Digests were re-suspended in 20 μL of 2% acetonitrile/0.2% formic acid and analysed using a Dionex Ultimate 3000 RSLC system coupled to a Sciex 6600 TripleTOF mass spectrometer. Peptides were first de-salted on an Acclaim PepMap C18 trap column (100 μm × 2 cm) for 2 min at 15 μL/min using 2% acetonitrile/0.2% formic acid, and then separated on an Acclain Pepmap C18 RSLC column (300 μm × 15 cm, 2 μm particle size). Peptide elution was achieved using a flow rate of 8 μL/min with a gradient of 4–60% B in 15 min (A: 0.1% formic acid; B: 80% acetonitrile/0.1% formic acid). An electrospray voltage of 5.5 kV was applied to the emitter. The 6600 TripleTOF mass spectrometer was operated in Data Dependant Acquisition mode. Precursor MS scans were acquired from m/z 400–1500 using an accumulation time of 250 ms followed by 30 MS-MS scans, acquired from m/z 100–1800 at 100 ms each, for a total scan of 3.3 s. Multiply charged ions (2+, 5+, 400–1500 m/z) were automatically fragmented in Q2 collision cells using nitrogen as the collision gas. Collision energies were chosen automatically as a function of m/z and charge.
Protein pilot v5.01.10 using Paragom search engine (Sciex) was used for comparison of the obtained MS/MS spectra against protein sequences in a Uniprot Swissprot 2016 database. Proteins with a threshold above ≥ 99.9% confidence were reported. Peptides with ≥ 95% confidence were used to report sequence coverage.
Results
Induction studies
Cultures of the strains R. rhodochrous ATCC BAA-870, R. rhodochrous BD-1780 and P. simplex BD-1781 were suspended in a minimal medium and induced with either benzonitrile (BZN) or dimethylformamide (DMF). After incubation for set periods, 100 mg wet wt cell paste of these cultures was used for a 1 h biocatalytic hydrolysis of 4.16 mg 3-cyanopyridine (3-CP; Fig. 2), and the products were identified and quantified by LC-MSMS and ultra-performance liquid chromatography mass spectrometry (UPLC-MS) in ESI+ mode (Fig. S2).

3-Cyanopyridine (1), nicotinamide (2) and nicotinic acid (3), dimethylformamide (4), benzonitrile (5) and ɛ-caprolactam
For all three strains, induction with DMF resulted in the production of nicotinic acid from 3-CP. In the absence of DMF, no nicotinic acid was formed whatsoever. Control cultures and BZN-induced cells of strains BD-1780 and BD-1781 generated only small amounts of nicotinamide, with large amounts of unreacted 3-CP remaining. However, for strain BAA-870, both control and BZN-induced cells showed complete conversion of 3-CP to nicotinamide (Fig. 3).

Biocatalytic hydrolysis of 3-cyanopyridine. 3-Cyanopyridine (blue); nicotinamide (red); nicotinic acid (green). X-axis: induction period: 0 h (1); 24 h (2); 48 h (3) and 72 h (4). No analysis was made of BAA-870 cells at 24 h. All reactions were carried out for 1 h in 1 mL with 100 mg dry wt cells and agitation at 28 °C in the presence of 40 mmol/L 3-CP. Average of six data points, consisting of triplicate flasks with duplicate samples per flask
Biocatalytic reactions
To generate sufficient product for complementary analytical procedures (chromatography and NMR), larger reactions were performed (200 mg wet wt biocatalyst and 75 mg 3-CP in 5 mL for 15 min). These revealed that the biocatalytic reactions of DMF-induced strain BD-1780 and BD-1781 cells generated only nicotinic acid after complete conversion of 3-CP. However, the DMF-induced BAA-870 cells also generated nicotinamide in addition to nicotinic acid products in these more concentrated reactions, possibly due to greater substrate availability for the nitrile hydratase. The product identity was confirmed using 1H and 13C NMR (Fig. S1; Table S3) as well as HPLC-MS. The isolated nitrile, amide and acid yields for each reaction were determined by weighing after chromatographic separation (Tables 1 and S4). The specific nitrilase activity of strains BD-1780 and BD-1781 on 3-cyanopyridine was calculated as 0.164 and 0.202 μmol/min/mg DCW, respectively.
High enantiomeric excess of a product of biocatalytic hydrolysis of a racemate is indicative of a single enzyme system and thus enantioselectivity of the biocatalysts was investigated. Reaction of racemic 3-amino-3-phenylpropanenitrile and 3-amino-3-(4-methoxyphenyl)propanenitrile in the presence of DMF-induced cells of strains BD-1780 and BD-1781 resulted in enantioselective kinetic resolution of these substrates. Excellent enantiomeric excess of the residual nitrile was observed when cells of either strain were reacted with 3-amino-3-phenylpropanenitrile (> 99% ee), while moderate enantioselectivity was found in the case of 3-amino-3-(4-methoxyphenyl)propanenitrile (34–41%) (Table S5). Based on the previously determined formation of (R)-3-amino-3-phenylpropionic acid by these strains (Chhiba et al. 2014), the residual nitrile can tentatively be assigned as the (S)-enantiomer (Fig. 4). In comparison, the nitrile hydratase of BAA-870 enantioselectively hydrolysed the opposite (S)-enantiomer of 3-amino-3-(4-methoxyphenyl)propanenitrile to generate the (S)-amide, but was relatively non-enantioselective in the hydrolysis of 3-amino-3-phenylpropanenitrile (Chhiba et al. 2012).

Enzymatic kinetic resolution of racemic β-aminonitriles
Enzyme inhibition studies
The enzyme system responsible (nitrilase or nitrile hydratase-amidase) could not be definitively assigned solely by the reaction product profile as evidenced by the differences seen between small scale and large scale reactions. Consequently, reactions with DMF-induced cells were subjected to nitrilase, nitrile hydratase and amidase inhibitors (Table 2).
Nicotinic acid generation by strains BD-1780 and BD-1781 was considerably reduced in the presence of the nitrilase inhibitors benzaldehyde (83–90%) and benzylamine (69–82%) (Table 2), indicating that nicotinic acid production was dependent on nitrilase activity. Nicotinic acid production by BAA-870 was influenced to a lesser extent (9–22%), presumably due to the presence of an alternative route via nitrile hydratase (as demonstrated in the induction studies) and an amidase (detected in molecular studies below). Conversely, the nitrile hydratase and amidase inhibitors had a negligible effect, indicating that the nitrilase route was dominant, even in the case of BAA-870. Both benzaldehyde and benzylamine resemble nitrilase reaction intermediates, with benzylamine similar to the thio-imidate intermediate and benzaldehyde analogous to the thio-ester intermediate, and hence, a model for competitive inhibition can be proposed (Fig. 5).

Proposed competitive inhibition points for benzaldehyde and benzylamine in the nitrilase catalytic cycle based on structural similarity to the reaction intermediates
Molecular characterisation
Inhibition studies undertaken on the DMF-induced cells suggested that the nitrile degrading activity of strains BD-1780 and BD-1781 was the result of an induced nitrilase enzyme. This, when coupled with the isolation of mixed amide and acid products from the DMF-induced cells of strain BAA-870, indicates that the nicotinic acid observed for BAA-870 could have been the result of an interplay of nitrilase, nitrile hydratase and amidase. Hence, it was decided to confirm the identity of the responsible enzymes though enzyme expression studies.
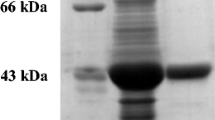
SDS-PAGE protein profiling was done on control, BZN and DMF-induced cells of all three strains (Fig. 6). With DMF induction, a dominant 40 kDa size protein, representing 11–12% of the total cytosolic soluble protein, was expressed in each case [BAA-870 (Fig. 6a lanes 11–12); strain BD-1780 (Fig. 6b lanes 6–7); strain BD-1780 (Fig. 6c lanes 6–7)]. No such band was observed in the control and BZN-induced cells. BAA-870 expressed an additional major protein at 25 kDa (Fig. 6a lanes 7–12).

SDS-PAGE gel of protein soluble fraction from induced and non-induced cells. BAA-870 (a): lane 1, PageRuler Plus Prestained Protein ladder; lanes 3–6, BSA standard concentrations 100–500 ng μL protein; lanes 7–8, BAA-870 control cell sample; lanes 9–10, BAA-870 BZN-induced cell sample; lanes 11–12, BAA-870 DMF-induced cell sample. Strain BD-1780 (b): lanes 2–3, BD-1780 control cell sample; lanes 4–5, BD-1780 BZN-induced cell sample; lanes 6–7, DMF-induced cell sample. Strain BD-1781 (c): lane 1, PageRuler; lanes 2–3, BD-1781 control cell sample; lanes 4–5, BD-1781 BZN-induced cell sample; lanes 6–7, DMF-induced cell sample
Protein bands correlating to 100, 70, 55, 40 and 25 kDa were excised and analysed by LC-MSMS. The SDS-PAGE protein profiles for P. simplex strain BD-1781 cells showed smearing consistent with extensive proteolysis, with only faint bands discernable even with addition of a protease inhibitor cocktail during cell lysis, so bands were excised relative to the molecular weight markers. Reference to the MEROPS website showed that P. simplex cells produce an array of proteases, with around 82 known and putative proteolytic enzymes identified in this one strain (MEROPS the Peptidase Database – EMBL-EBI 2017), which may explain the general protein degradation.
Nitrilase peptide fragments corresponding to Uniprot Q-03217 were found in the 40 kDa protein band from DMF-induced cells of all three strains evaluated, with no nitrilase fragments being found in any of the control or BZN-induced cells. The protein fragments showed > 99% sequence identity to an aliphatic nitrilase protein produced by R. rhodochrous J1 with 36.1% of protein coverage observed in BAA-870; 92.62% of the total sequence coverage located in the BD-1780; and 35.3% sequence coverage identified in the strain BD-1781 sample (Fig. S3).
The 25 kDa band in all the SDS-PAGE lanes of the cell lysates of BAA-870 cells was determined to be the alpha subunit of a constitutively expressed low molecular weight nitrile hydratase (Fig. S4). An amidase was also identified in all cell lysates of BAA-870 (Fig. S5) as the 55 kDa band (P-84650, enantioselective amidase from R. rhodochrous, 6% sequence coverage, > 99% sequence similarity) (Pertsovich et al. 2005). No nitrile hydratase or amidase protein sequence fragments were located in any of the strain BD-1780 and BD-1781 samples, suggesting the inducible nature of these enzymes in these strains. The characterisation of the three strains under DMF induction has been summarised in Table 3).
An array of transcription and translational proteins were found to be present in the lysates of the DMF-induced cells. These included both the 30S and 50S ribosomal proteins crucial for protein translation; DNA-directed RNA polymerase; various elongation factors (EF-U, EF-G as well as EF-Ts); as well as numerous 60 kDa chaperonin proteins. One chaperonin peptide identified in all three strains was Uniprot Q-2JA12 (Fig. S6), which showed high amino acid sequence similarity to Uniprot O50305 (GroEL 60 kDa chaperonin protein from Bacillus halodurans), which is associated with enhanced nitrilase activity (Petříčková et al. 2012).
Discussion
DMF has not previously been assessed as a nitrilase inducer. Although organic solvents, including DMF, have previously been used to enhance nitrilase and nitrile hydratase activity in biocatalytic reactions (Stevenson et al. 1992; Nagasawa et al. 2000; Kaul and Banerjee 2008; Tauber et al. 2000), these effects were most likely related to solubility of the enzyme substrate during the reaction. In the present study, it was shown that Rhodococcal strains BD-1780 and BAA-870 and the closely related P. simplex strain BD-1781 expressed a DMF-inducible nitrilase.
DMF-induced cells of strains BD-1780 and BD-1781 were shown to convert 3-cyanopyridine (3-CP) solely to nicotinic acid. Addition of the nitrilase inhibitors benzaldehyde and benzylamine reduced 3-CP hydrolysis in agreement with Stevenson et al. (1992) who showed that these compounds competitively inhibited nitrilase activity in resting cells of Rhodococcus ATCC 39484. The results of biocatalysis and enzyme inhibition studies were confirmed by SDS-PAGE and LC-MSMS amino acid sequence analysis which showed that DMF-induced cells expressed a dominant ~ 40 kDa protein with a sequence identical to a known nitrilase. BAA-870 showed additional constitutive expression of a nitrile hydratase that synthesised nicotinamide. Inhibitors for nitrile hydratase and amidase (Yamada and Kobayashi 1996; Endo and Watanabe 1989; Prasad et al. 2009; Nagasawa et al. 1991; Tani et al. 1989; Zangi et al. 2009; Precigou et al. 2004; Leonova et al. 2000; Bauer et al. 1998; Brady et al. 2004) had negligible effect, indicating that nitrilase was the primary nicotinic acid producer.
Nitrilase enzyme activation by subunit dimerization and subsequent polymerisation is often achieved through the action of specialised chaperone proteins. Nitrilase purification studies on Bacillus pallidus Dac521, Comomonas testosteroni and Pseudomonas fluorescens DSM 7155 showed that nitrilases are often co-purified with the chaperonin GroEL (Lévi-Schil et al. 1995; Almatawah et al. 1999; Layh et al. 1998). Similar enhancement of activity was seen by co-expressing fungal nitrilases from Gibberella moniliformis and Penicillium marneffei with GroEL (Petříčková et al. 2012). In the present study, GroEL was co-expressed with nitrilase in strains BD-1780, BD-1781 and BAA-870.
The role that DMF plays in nitrilase induction in these cells is not obvious but, as DMF is toxic to most cells, nitrilase activity induction may be part of a cell detoxification mechanism. Nitrilase expression in plants is often to facilitate cellular detoxification when the breakdown of glucosinolates yields toxic isothiocyanates and nitrile compounds (Miller and Conn 1980). DMF (or a metabolite such as dimethylamine or N-methylformamide) could function as a nitR gene regulator. Nitriles, although the most common, are not the only inducers of nitrilase (Table S2). Herai et al. (2004) have demonstrated that a ε-caprolactam-induced R. rhodochrous J1 nitrilase expression system (Nagasawa et al. 1990) could be of value in industrial heterologous protein production. By superimposing ɛ-caprolactam and DMF, a structural similarity is evident (Fig. 7), implying that the mechanism of induction may be similar. However, DMF is commercially attractive at a fraction of the cost of ε-caprolactam. Preliminary results (SDS-PAGE) from our laboratory indicate that the structurally similar N,N-dimethylacetamide and N-methylpyrrolidone also induce nitrilase in strain BD-1780.

Overlay after energy minimisation of DMF and ɛ-caprolactam (ChemDraw) showing the structural smilarities of the two molecules
In conclusion, DMF is a novel inducer that can be used to selectively induce nitrilase but not nitrile hydratase. This permits the catalytic specificity inherent in the nitrilase to be expressed, unencumbered by the competing nitrile hydratase-amidase system. This is particularly desirable when resolving enantiomers or isomers as the competing nitrile biocatalytic systems may have opposing enantio- or regio-selectivity, potentially resulting in poor enantiomeric excess (ee) or isomer yields. Through application of multiple analytical techniques, we were also able to unequivocally define which enzymes were responsible for biocatalytic nitrile hydrolysis and enantiomeric resolution in prior studies using these microbial strains (Chhiba et al. 2012, 2014; Brady et al. 2004; Rapheeha et al. 2017; Kinfe et al. 2009). These results expand the small group of non-nitrile nitrilase inducers previously only represented by the lactams reported by Nagasawa et al. (1990).
References
Ahmed K, Kumar M, MS KCG, Shaik TB (2011) Bioconversion of acrylonitrile to acrylic acid by Rhodococcus ruber strain AKSH-84. J Microbiol Biotechnol 21:37–42
Almatawah QA, Cramp R, Cowan DA (1999) Characterization of an inducible nitrilase from a thermophilic bacillus. Extremophiles 3:283–291
Badoei-Dalfard A, Karami Z, Ramezani-pour N (2016) Bench scale production of nicotinic acid using a newly isolated Stenotrophomonas maltophilia AC21 producing highly-inducible and versatile nitrilase. J Mol Catal B Enzym 133:S552–S559
Bauer R, Knackmuss H-J, Stolz A (1998) Enantioselective hydration of 2-arylpropionitriles by a nitrile hydratase from Agrobacterium tumefaciens strain d 3. Appl Microbiol Biotechnol 49:89–95
Binfeng L, Jinhuan S, Tao J (2011) Enzyme and process development for production of nicotinamide. Org Process Res Dev 15:291–293
Borsook H, Davenport HW, Jeffreys CEP, Warner RC (1937) The oxidation of ascorbic acid and its reduction in vitro and in vivo. J Biol Chem 117:237–279
Brady D, Beeton A, Kgaje C, Zeevaart J, van Rantwijk F, Sheldon RA (2004) Characterisation of nitrilase and nitrile hydratase biocatalytic systems. Appl Microbiol Biotechnol 64:76–85
Brenner C (2002) Catalysis in the nitrilase superfamily. Curr Opin Struct Biol 12:775–782
Chhiba VP, Bode ML, Mathiba K, Kwezi W, Brady D (2012) Enantiomeric biocatalytic hydrolysis of β-aminonitriles to β-aminoamides using Rhodococcus rhodochrous ATCC BAA-870. J Mol Catal B Enzym 76:68–74
Chhiba VP, Bode M, Mathiba K, Brady D (2014) Enzymatic stereoselective synthesis of β-amino acids. In: Riva S, Fessner WD (eds) Cascade biocatalysis: integrating stereoselective and environmentally friendly reactions. Wiley-VCH, Germany, pp 297–313
Cramp RA, Cowan DA (1999) Molecular characterization of a novel thermophilic nitrile hydratase. Biochem Biophys Acta 1431:249–260
Endo T, Watanabe I (1989) Nitrile hydratase of Rhodococcus sp. N774 purification and amino acid sequences. FEBS Lett 243:61–64
Fang S, An X, Liu H, Cheng Y, Hou N, Feng L, Huang X, Li C (2015) Enzymatic degradation of aliphatic nitriles by Rhodococcus rhodochrous BX2, a versatile nitrile-degrading bacterium. Bioresour Technol 185:28–34
Fernandes BCM, Mateo C, Kiziak C, Chmura A, Wacker J, van Rantwijk F, Stolz A, Sheldon RA (2006) Nitrile hydratase activity of a recombinant nitrilase. Adv Synth Catal 348:2597–2603
Goncalves ER, Hara H, Miyazawa D, Davies JE, Eltis LP, Mohn WW (2006) Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Appl Environ Microbiol 72:6183–6193
Gong J-S, Lu Z-M, Li H, Shi JS, Zhou Z-M, Xu Z-H (2012) Nitrilases in nitrile biocatalysis: recent progress and forthcoming research. Microb Cell Factories 11:142
Gradley ML, Deverson CJF, Knowles CJ (1994) Asymmetric hydrolysis of R-(-)-S (+)-2-methylbutyronitrile by Rhodococcus rhodochrous NCIMB 11216. Arch Microbiol 161:246–251
Gregoriou M, Brown PR (1979) Inhibition of the aliphatic amidase from Pseudomonas aeruginosa by urea and related compounds. Eur J Biochem 96:101–108
Harper DB (1977) Microbial metabolism of aromatic nitriles: enzymology of aromatic nitriles: enzymology of C-N cleavage by Nocardia sp. (Rhodococcus group) N. C. I. B. 11216. Biochem J 165:309–319
He Y-C, Wu Y-D, Pan X-H, Ma C-L (2014) Biosynthesis of terephthalic acid, isophthalic acid and their derivatives from the corresponding dinitriles by tetrachloroterephthalonitrile-induced Rhodococcus sp. Biotechnol Lett 36:341–347
Herai S, Hashimoto Y, Higashibata H, Maseda H, Ikeda H, Omura S, Kobayashi M (2004) Hyper-inducible expression system for Streptomycetes. Proc Natl Acad Sci 101:14031–14035
Kaul P, Banerjee UC (2008) Predicting enzyme behaviour in nonconventional media: correlating nitrilase function with solvent properties. J Ind Microbiol Biotechnol 35:713–720
Kinfe HH, Chhiba V, Frederick J, Bode ML, Mathiba K, Steenkamp PA, Brady D (2009) Enantioselective hydrolysis of β-hydroxy nitriles using the whole cell biocatalyst Rhodococcus rhodochrous ATCC BAA-870. J Mol Catal B Enzym 59:231–236
Kobayashi M, Nagasawa T, Yamada H (1989) Nitrilase of Rhodococcus rhodochrous J1. Eur J Biochem 182:349–356
Kobayashi M, Goda M, Shimizu S (1998) The catalytic mechanism of amidase also involves nitrile hydrolysis. FEBS Lett 439:325–328
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Layh N, Parratt J, Willetts A (1998) Characterization and partial purification of an enantioselective arylacetonitrilase from Pseudomonas fluorescens DSM 7155. J Mol Catal B Enzym 5:476–474
Leonova TE, Astaurova OB, Ryabchenko LE, Yanenko AS (2000) Nitrile hydratase of Rhodococcus: optimization of synthesis in cells and industrial applications for acrylamide production. Appl Biochem Biotechnol 88:231–241
Lévi-Schil S, Soubrier F, Crutz-Le Coq A-M, Faucher D, Crouzet J, Pétré D (1995) Aliphatic nitrilase from a soil-isolated Comomonas testosteroni sp.: gene cloning and overexpression, purification and primary structure. Gene 161:15–20
Martínková L, Uhnáková B, Pátek M, Nešvera J, Křen V (2009) Biodegradation potential of the genus Rhodococcus. Environ Int 35:162–177
Martínková L, Rucká L, Nešvera J, Pátek M (2017) Recent advances and challenges in the heterologous production of microbial nitrilases for biocatalytic applications. World J Microbiol Biotechnol 33:8
Mateo C, Chmura A, Rustler S, van Rantwijk F, Stolz A, Sheldon RA (2006) Synthesis of enantiomerically pure (S)-mandelic acid using an oxynitrilase-nitrilase bienzymatic cascade: a nitrilase surprisingly shows nitrile hydratase activity. Tetrahedron Asymmetry 17:320–323
Miller JM, Conn EE (1980) Metabolism of hydrogen cyanide by higher plants. Physiol Plant 65:1199–1202
Nagasawa T, Ryuno K, Yamada H (1986) Nitrile hydratase of Brevibacterium R-312 purification and characterisation. Biochem Biophys Res Commun 139:1304–1312
Nagasawa T, Nakamura T, Yamada H (1990) ε-Caprolactam, a new powerful inducer for the formation of Rhodococcus rhodochrous J1 nitrilase. Arch Microbiol 155:13–17
Nagasawa T, Takeuchi K, Yamda H (1991) Characterization of a new cobalt-containing nitrile hydratase purified from urea-induced cells of Rhodococcus rhodochrous J1. Eur J Biochem 196:581–589
Nagasawa T, Weiser M, Nakamura T, Iwahara H, Yoshida T, Gekko K (2000) Nitrilase or Rhodococcus rhodochrous J1. Conversion into the active form by subunit association. Eur J Biochem 267:138–144
Nageshwar YVD, Sheelu G, Shambu RR, Muluka H, Mehdi N, Malik MS, Kamal A (2011) Optimization of nitrilase production from Alcaligenes faecalis MTCC 10757 (IICT-A3): effect of inducers on substrate specificity. Bioprocess Biosyst Eng 34:515–523
Pertsovich SI, Guranda DT, Podchernyaev DA, Yanenko AS, Svedas VK (2005) Aliphatic amidase from Rhodococcus rhodochrous M8 is related to the nitrilase/cyanide hydratase family. Biochem Mosc 70:1280–1287
Petříčková A, Veselá AB, Kaplan O, Kubáč D, Uhnáková B, Malandra A, Felsberg J, Rinágelová A, Weyrauch P, Křen V, Bezouška K, Martínková L (2012) Purification and characterization of heterologously expressed nitrilases from filamentous fungi. Appl Microbiol Biotechnol 93:1553–1561
Prasad S, Misra A, Jangir VP, Awasthi A, Raj J, Bhalla TC (2007) A propionitrile-induced nitrilase of Rhodococcus sp. NDB 1165 and its application in nicotinic acid synthesis. World J Microbiol Biotechnol 23:345–353
Prasad S, Raj J, Bhalla TC (2009) Purification of a hyperactive nitrile hydratase from resting cells of Rhodococus rhodochrous PA-34. Indian J Microbiol 49:237–242
Precigou S, Wieser M, Pommares P, Goulas P, Duran R (2004) Rhodococcus pyridinovorans MW3, a bacterium producing a nitrile hydratase. Biotechnol Lett 26:1379–1384
Rapheeha OK, Roux-van der Merwe MP, Badenhorst J, Chhiba V, Bode ML, Mathiba K, Brady D (2017) Hydrolysis of nitriles by soil bacteria: variation with soil origin. J Appl Microbiol 122:686–697
Robinson WG, Hook RH (1964) Ricinine nitrilase: I. Reaction producer and substrate specificity. J Biol Chem 239:4257–4262
Sharma NN, Sharma M, Bhalla TC (2011) An improved nitrilase-mediated bioprocess for synthesis of nicotinic acid from 3-cyanopyridine with hyperinduced Nocardia globerula NHB-2. J Ind Microbiol Biotechnol 38:1235–1243
Stalker DM, Malyj LB, McBride KE (1988) Properties of a nitrilase specific for the herbicide bromoxynil and corresponding nucleotide sequence analysis of the bxn gene. J Biol Chem 263:6310–6314
Stevenson DE, Feng R, Dumas F, Groleau D, Mihoc A, Storer AC (1992) Mechanistic and structural studies on Rhodococcus ATCC 39484 nitrilase. Biotechnol Appl Biochem 15:283–302
Tani Y, Kurihara M, Nishise H (1989) Characterization of nitrile hydratase and amidase, which are responsible for the conversion of dinitriles to mononitriles, from Corynebacterium sp. Agric Biol Chem 53:3151–3158
Tauber MM, Cavaco-Paulo A, Robra K, Gubitz GM (2000) Nitrile hydratase and amidase from Rhodococcus rhodochrous hydrolyze acrylic fibers and granular polyacrylonitriles. Appl Environ Microbiol 66:1634–1638
Thalenfeld B, Grossowicz N (1976) Regulatory properties of an inducible aliphatic amidase in a thermophilic Bacillus. J Gen Microbiol 94:131–141
Thuku RN, Brady D, Benedik MJ, Sewell BT (2009) Microbial nitrilases: versatile, spiral forming enzymes. J Appl Microbiol 106:703–727
Vaudel M, Burkhart JM, Zahedi RP, Oveland E, Berven FS, Sickmann A, Martens L, Barsnes H (2015) PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat Biotechnol 33:22–24
Vejvoda V, Kubác D, Davidová A, Kaplan O, Śulc M, Śveda O, Chaloupková R, Martínkova L (2010) Purification and characterization of nitrilase from Fusarium solani IMI96840. Process Biochem 45:1115–1120
Yamada H, Kobayashi M (1996) Nitrile hydratase and its application to industrial production of acrylamide. Biosci Biotechnol Biochem 60:1391–1400
Yusuf F, Chaubey A, Jamwal U, Parshad R (2013) A new isolate from Fusarium proliferatum (AUF-2) for efficient nitrilase production. Appl Biochem Biotechnol 171:1022–1031
Zangi R, Zhou R, Berne BJ (2009) Urea’s action on hydrophobic interactions. J Am Chem Soc 131:1535–1541
Zhang ZJ, Xu JH, He YC, Ouyang LM, Liu YY (2011) Cloning and biochemical properties of a highly thermostable and enantioselective nitrilase from Alcaligenes sp. ECU0401 and its potential for (R)-(-)-mandelic acid production. Bioprocess Biosyst Eng 34:315–322
Zheng JG, Chen J, Liu ZQ, Wu MH, Xing LY, Shen YC (2008) Isolation, identification and characterization of Bacillus subtilis ZJB-063, a versatile nitrile-converting bacterium. Appl Microbiol Biotechnol 77:985–993
Websites
Merops-the peptidase database 2017, accessed 20th March 2017, <https://www.ebi.ac.uk/merops/cgi-bin/speccards?sp=sp034913;type=peptidase>
Acknowledgements
We would like to thank the following people at the University of the Witwatersrand: Prof. Joseph Michael; Prof. Charles De Koning; and Dr. Amanda Rousseau. At the CSIR, we would like to extend thanks to Mr. Sipho Mamputhu (for assistance with LC-MSMS protein sequencing) as well as Dr. Priyen Pillay, Dr. Lusisiwe Kwezi, Dr. Ofense Poe, Advaita Singh, Sibongile Mtimka, Dr. Daria Rutkovska, Dr. Kevin Wellington and Dr. Tsepo Tsekoa.
Funding
Support was received from the Department of Science and Technology Biocatalysis Initiative (Grant 0175/2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 649 kb)
Rights and permissions
About this article

Cite this article
Chhiba-Govindjee, V.P., Mathiba, K., van der Westhuyzen, C.W. et al. Dimethylformamide is a novel nitrilase inducer in Rhodococcus rhodochrous. Appl Microbiol Biotechnol 102, 10055–10065 (2018). https://doi.org/10.1007/s00253-018-9367-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9367-9