
Abstract
The incidence of shrimp disease is closely associated with the microbial composition in surrounding water, but it remains uncertain whether microbial indicator phylotypes are predictive for shrimp health status (healthy or diseased). To test this idea, we combined the data from our previous works, to investigate the feasibility of indicator phylotypes as independent variables to predict the health status during a shrimp culture procedure. The results showed linearly increased dissimilarities (P < 0.001) of the bacterioplankton community over time, while the communities dramatically deviated from this defined trend when disease occurred. This sudden shift in the bacterial community appears to cause severe mass mortality of the shrimps. In particular, we created a model to identify indicators that discriminated ponds with diseased shrimp populations from these with healthy shrimp populations. As a result, 13 indicative families were screened, in which seven are healthy indicator and six are diseased indictor. An improved logistic regression model additionally revealed that the occurrences of these indicator families could be predictive of the shrimp health status with a high degree of accuracy (>79 %). Overall, this study provides solid evidences that indicator phylotypes could be served as independent variables for predicting the incidences of shrimp disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
To sustain a high production, the feed supply generally exceeds the required level in shrimp culture ponds. This practice causes a gradual deterioration of water quality, particularly in the mid- to late periods of shrimp cultivation (Sugiura et al. 2006; Ma et al. 2013). The deteriorated water quality exerts strong pressures on shrimp health, often leading to severe disease outbreaks and economic losses, which has become a major threat to aquaculture (Defoirdt et al. 2011; Ferreira et al. 2011). For these reasons, it is urgently needed to establish reliable strategies for forecasting the incidences of shrimp disease. Considering the innumerable and complicated variations in water parameters, monitoring multiple geochemical factors has been wildly employed to generate water quality indices (Ferreira et al. 2011; Ma et al. 2013) for disease forecasting. However, in practice, it is generally difficult to set a threshold value for a specific abiotic parameter to evaluate the disease risk precisely. Thus, the application of only traditional physicochemical variables to assess the shrimp health status may be inadequate.
Recently, ample evidences have shown that there is an inextricable correlation between plankton community and shrimp health, with distinct microbial communities between ponds with healthy shrimp populations (PHS) and diseased ones (PDS) (Lucas et al. 2010; Boutin et al. 2013; Zhang et al. 2014). Thus, the microbial assemblages that preferably occurred in PHS or PDS may serve as diagnostic species or indicator species (a concept initially referred in macroecology, McGeogh 1998; Chytrý et al. 2002), for indicating the incidences of shrimp disease. Recently, the determination indicator of species has become an active research topic in microbial ecology (Paerl et al. 2003; Berry et al. 2012; Fortunato et al. 2013; Xiong et al. 2014a), which has provided solid evidences that microbial indicators were strongly associated with health status or habitat condition. For example, Berry et al. (2012) revealed that shifts in the bacterial intra-family structure can be indicative for dextran sodium sulfate (DSS)-induced disease. Similarly, Fortunato et al. (2013) showed that indicator taxa characterized the dynamics of seasonal condition. However, these findings center on the discriminative pattern by indicator taxa between groups (i.e., healthy or diseased, disturbance states), rather than prediction over time (De Cáceres and Legendre 2009; Hilbe 2009; Zhang et al. 2014). In other words, to date, there is no model-based approach to predict the incidence of shrimp disease on temporal scale. Consequently, it is unclear whether indicator phylotypes could be used as independent variables for predicting the health status over an entire duration of shrimp culture.
Indeed, we have found that the bacterioplankton communities could discriminate PHS from PDS (Zhang et al. 2014) and that bacterial temporal dynamics are predictable to a certain extent (Xiong et al. 2014b). These findings address the idea to apply bacterial indicator phylotypes for predicting health status (healthy or diseased) during shrimp cultivation. It is known that stressful conditions such as those occurring in ponds induce changes in bacterial community, thus enhancing the virulence of opportunistic pathogens to cause shrimp disease (Boutin et al. 2013; Zhou et al. 2012). We, therefore, speculate that dramatically shifts in bacterial community would cause the occurrences of shrimp disease; thus, the indicator phylotypes could provide a warning of the increased risk of disease outbreak. However, this idea has never been tested directly by experimental data in aquaculture ponds. To test this hypothesis, we collected bacterial community data from our previous works (Xiong et al. 2014b; Zhang et al. 2014) and created a model to screen indicator phylotypes at different taxonomic levels. Further, we applied an improved logistic regression model (Hilbe 2009; De Cáceres et al. 2010), based on the occurrence of indicator phylotypes to predict the health status over duration of shrimp farming, rather than a simple t test to discriminate ponds with healthy and diseased shrimps (Zhang et al. 2014). To our knowledge, this is the first study that shows evidence that the indicator bacterioplankton assemblages are reliable for predicting the incidence of shrimp disease.
Materials and methods
Experimental design and water sample collection
The shrimp ponds investigated in this study are located at the Zhanqi, Ningbo, eastern China (29° 32′ N, 121° 31′ E). These 30 ponds are approximately the same size (2,000 m2) and are identically managed. Shrimp (Litopenaeus vannamei) juveniles were introduced into the ponds on 25 March, 2012. The surface water samples (25–50 cm) were collected at various time points separated by 7 to 10 days (over a span of 42 days, from 29 April to 10 June) in six selected ponds, corresponding to 35, 45, 55, 63, 69, and 77 days after shrimp inoculation. This duration covers the periods during which the risk of a disease outbreak was high (58 ± 8 days after the introduction of juvenile shrimp) (Lemonnier et al. 2006). Consistently, a disease outbreak caused massive mortality of shrimp in six ponds (independent of our monitored ponds) on 27 May, 63 days after inoculation. Farmers exchanged 80 % water and applied antibiotics in disease ponds. We designed to take samples from diseased ponds over time, while the shrimps were harvested on day 80, and additional sampling was stopped. We collected water samples from these six ponds with disease shrimp populations. Water samples were stored at 4 °C during transportation to the lab.
Data collection and deposition
Water biogeochemical variables and bacterial 16S rRNA gene (V4-V5 region) pyrosequencing data were collected from our previous works (Xiong et al. 2014b; Zhang et al. 2014). See abiotic information in Table S1. The combined sequences data were deposited in the DDBJ (http://www.ddbj.nig.ac.jp/) Sequence Read Archive and are available under the accession number DRA001853.
Statistical analysis
The sequence reads were low in eight samples from ponds with healthy shrimps, which were entirely removed for further analysis. Nonmetric multidimensional scaling (NMDS) analysis was implemented to evaluate the overall differences of microbial community structure to determine changes in beta diversity (Clarke, 1993). Parametric permutational multivariate analysis of variance (perMANOVA) was conducted to quantitatively evaluate the effects of disease and sampling time on the composition of microbial community using the “adonis” function (Anderson 2001). A permuted multivariate analysis of variance (MANOVA) was used to disentangle the effects of disease and sampling time on the variations at phyla level in R v.2.11.0 with the “vegan” package (www.R-project.org).
Indicator phylotype identification and prediction model construction
In total, 34 samples were collected in this study, with six samples from PDS and 28 samples from PHS. We created a model to identify the indicator phylotypes that efficiently discriminate the bacterioplankton composition from PHS to PDS. The temporal dynamics of bacterial communities in shrimp culture ponds (Xiong et al. 2014b); we used the samples from healthy and diseased ponds at D63 to minimize unequal sampling size and then applied the indicative assemblages to predict the health status across the 34 samples. The match rate of test sample to modeling sample is the predictive accuracy. Bacterial communities were analyzed at different taxon levels that can be, for instance, genus, family, or class level. The community tables at different taxon levels were generated during Qiime workflow (Caporaso et al. 2010). The indicator phylotypes analysis determines the strength of the association between a given taxon and its target condition (De Cáceres and Legendre 2009).
-
1.
Basic principles
-
(a)
The sum of the relative abundances of indicator phylotypes (hereafter called “SRAIP”) for a given sample is employed as an indicator to evaluate the health status (healthy or diseased) of shrimp populations at corresponding pond;
-
(b)
The inner-group variation coefficient of SRAIP should be as small as possible, that is, the SRAIP is relatively stable for a specified group (PHS or PDS);
-
(c)
The inter-group differences of SRAIP should be as great as possible, i.e., the difference is obvious enough to distinguish the groups;
-
(d)
The relative abundance of each characteristic phylotype should be relatively high for practical detection purpose.
-
(a)
-
2.
Algorithm
-
(a)
Given that there are n samples (i = 1, 2…, n), and m phylotypes (j = 1, 2…, m), which were separated into two groups, bacterial communities from PHS or PDS, calculate the mean relative abundance of phylotype j in each group, that is, ad j and ah j ;
-
(b)
If ad j > ah j (had an arithmetic mean difference of 0.5 % relative abundance between PHS and PDS), phylotype j is initially defined as quasi-diseased community; conversely, defined as quasi-healthy community. As a result, we obtain m d quasi-diseased phylotypes and m h quasi-healthy phylotypes;
-
(c)
For all the m d quasi-diseased phylotypes, remove phylotype j and calculate the sum of the relative abundances (s ij ) of the residual quasi-diseased phylotypes for each sample. Using s ij to estimate the significant difference (p jd ) between PHS and PDS based on t test;
-
(d)
If the p jd value is the lowest among the m d quasi-diseased phylotypes when phylotype j has been removed (that is, phylotype j contributes negligible differences between groups), phylotype j is deleted in subsequent analysis, thus generating a combination that contains m d -1 quasi-diseased phylotypes;
-
(e)
Repeat steps (c)–(d); each cycle removes one phylotype; the k-th step makes a combination comb x containing m d−k phylotypes. Repeat until m d−k reaches the pre-given minimum number x of diseased indicator phylotypes;
-
(f)
For the combination of x (comb x , x = m d−k ) from steps (c)–(e), calculate the sum of the relative abundance of the comb x , sd x , and sh x for each sample; the mean, ad x , and ah x ; the variation coefficient, vd x , and vh x of the comb x within the samples from PDS; and the PHS (each group with six samples on day 63), respectively. Using ad x and ah x to estimate the significant difference (P n ) between PDS and PHS based on t test;
-
(g)
Using sum of the relative abundance to classify all the samples into PDS or PHS, i.e., if the sd x of sample i greater than ad x , it is categorized into PDS; conversely, categorized into PHS. Calculate the accuracy rates, Rd x , and Rh x for PDS and PHS, respectively;
-
(h)
Specify the following parameters: the accuracy rate R for distinguishing PDS from PHS, the mean ad and the coefficient of variation vd of SRAIP, and the maximum number xd max and minimum number xd min of indicator phylotypes. According to those parameters and the results above, screening out the optimum combination of diseased phylotypes, named the diseased indicators (DI);
-
(i)
In steps (c)–(h), replacing the quasi-diseased phylotypes with quasi-healthy phylotypes to acquire the healthy indicators (HI) from the residual m - m d phylotypes.
-
(a)
-
3.
Prediction method
We used an improved logistic regression model to predict the shrimp health status with following formula (Hilbe 2009): \( P=\frac{ \exp \left({\beta}_0+{\beta}_1{X}_1+\dots +{\beta}_n{X}_n\right)}{1+ \exp \left({\beta}_0+{\beta}_1{X}_1+\dots +{\beta}_n{X}_n\right)} \), where P is the health status, X 1, X 2, …, X n (the relative abundances of the DI or HI) are independent variables, and β 1, β 2, …, β n are parameters for logistic regression model. P is the health status of sample, P = 1 for healthy and P = 0 for diseased ponds.
This model was used to predict the health status based on the occurrence of indicator phylotypes (independent variable) over the duration of shrimp culture. The match rate of test sample to modeling sample is the predictive accuracy.
Results
Distribution of taxa and phylotypes
Across all water samples, we collected a total of 241,689 high-quality sequences and 4,502–8,731 sequences per sample (mean = 7,108). We were able to classify 94.1 % of those sequences at the phylum level. The dominant phyla were Bacteroidetes, Alphaproteobacteria, Actinobacteria, Gammaproteobacteria, and Cyanobacteria (relative abundance > 5 %), representing more than 79 % of the bacterial sequences (Fig. S1). In addition, Acidobacteria, Chloroflexi, and Firmicutes were present in most samples at low abundance (data not shown). Particularly, the occurrences of several phyla significantly changed between ponds with healthy and diseased shrimps. Specifically, the relative abundances of Flavobacteria, Gammaproteobacteria, and Betaproteobacteria dramatically decreased at PDS relative to that of PHS; by contrast, those of unclassified bacteria, Planctomycetes and Gemmatimonadetes, displayed the opposite trend (Fig. S2).
Sudden shift in the bacterioplankton communities was associated with shrimp disease outbreak
Based on the detected operational taxonomic units (OTUs) across the samples, a NMDS ordination analysis clearly revealed a continuous succession of bacterioplankton assemblages during our monitored shrimp-farm procedure. At the diseased ponds, specifically, bacterial community structures were distinct from that of healthy ones, primarily separated by the second axis (Fig. 1a), although the community richness and diversities did not vary dramatically over time (Fig. S3). The linear function showed a significant correlation (P < 0.001) between NMDS axis 1 (as a proxy for the bacterial community dissimilarity) and sampling time, but that incremental dissimilarities in diseased ponds dramatically deviated from the trend (Fig. 1b). In particular, this marked shift in the bacterioplankton composition was associated with severe shrimp mortality. It seems that the bacterial communities between PDS and PHS were mainly separated by NMDS axis 2; thus, values of NMDS axis 2 were regressed against geochemical factors. There were significant correlations (P < 0.05) between community distances and TN, TP, or COD (Fig. S4), which is in concert with the notion that the stressful environmental parameters usually induce the virulence of shrimp disease (Boutin et al. 2013; Zhou et al. 2012).

Nonmetric multidimensional scaling (NMDS) plot derived from the Jaccard distances between water samples (a) with symbols coded by sampling time, and the first component from NMDS of the Jaccard distances regressed against sampling time using a linear function for the bacterial community (b). H healthy, D diseased
Note that the community composition did not vary significantly between consecutive sampling points, such as H45 vs. H55 and H63 vs. H69. However, the bacterioplankton assemblages were dramatically changed from PHS to each PDS (Table S2). This was still true when compared with bacterioplankton communities between PHS and PDS on the same sampling day (H63 vs. D63, P = 0.005, Table S2). Furthermore, we used a two-way ANOVA to evaluate the effects of sampling time and disease on the occurrences at phylum level and found that more phyla were affected by disease outbreak, including the predominant phyla of Bacteroidetes and Alphaproteobacteria (Table 1). Additionally, permutational multivariate analysis of variance (perMANOVA) showed that sampling time and disease outbreak significantly affected the bacterial community structure; specifically, sampling time (8.3 %, P = 0.001) exerted stronger effects on the dynamics of the bacterial community than disease outbreak did (6.1 %, P = 0.001). However, when the samples (before the emergences of shrimp disease) were stepwise deleted in the analysis, the relative importance of disease outbreak successively increased (Table 2), suggesting that disease outbreak is an independent factor that triggers the shifts in bacterial communities. Overall, the results demonstrated that the occurrence of disease was closely associated with shifts in bacterioplankton composition, concomitantly with a moderately temporal dynamics.
Model-based approach for predicting the shrimp health status
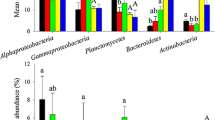
To determine if the degree of phylotype kinship influences predictive accuracy, we identified indicator phylotypes at different taxonomic levels (that is, at phylum, class, order, family, and genus levels) as proposed before (Berry et al. 2012). The occurrences of the indicator phylotypes were served as independent variables to evaluate the predictive accuracy by an improved logical regression model (Hilbe 2009). The results showed that family-level characterization revealed the highest predictive accuracy (Table 3). Therefore, a family-level indicator phylotype was employed for subsequent indication of the shrimp health status. Specifically, there were seven numerically dominant healthy indicator phylotypes; their relative abundances were substantially (P < 0.05) decreased at PDS (Fig. 2a). In contrast, six diseased indicator phylotypes were identified, whose relative abundances were significantly (P < 0.05) increased relative to those at PHS (Fig. 2b). Applying the healthy indicator phylotypes to predict the health status across the 34 samples, the accuracy was 88.0 % (the accuracy for PHS was 85.0 %, and PDS was 100 %, see detail in Table 3), while the predictive accuracy of diseased indicator phylotypes was 79.4 % (75.0 % for PHS and 100% for PDS). Notably, the predictive accuracy is higher at the mid- to later stages than that at the initial stage during shrimp farming, that is, the improperly predicted samples mainly originated in early stage samples (Fig. 3).

Bacterial indicator phylotypes of ponds with healthy and diseased shrimp populations. Indicator families are presented with their mean relative abundance ± standard deviation (N = 6) in water bodies with healthy or diseased shrimp populations. The mean relative abundances of specific family that significantly decreased (P < 0.05, unpaired t test) in ponds with diseased shrimp populations are designated by the term healthy indicator (a). The corresponding mean for the other families is designated by the term diseased indicator (b)

The predictive accuracy based on the occurrence of healthy indicator (HI) and diseased indicator (DI) for healthy samples over the duration of shrimp culture. If the health status of the test sample matches corresponding modeling sample, the accuracy is 1, otherwise, the accuracy is 0. Because of the accuracies for predicting diseased samples are 100 % by HI or DI (Table 2), the biplot is not shown
Strikingly, the regression between the sum of the relative abundance of diseased indicator phylotypes in a given sample with that of healthy indicator phylotypes was negatively correlated (r = −0.847, P < 0.001) across the samples (Fig. 4). Thus, these indicator phylotypes could be used to accurately estimate the shrimp health status, particularly for predicting shrimp disease outbreak.

The relationship between the sum of the relative abundances of healthy indicators and the sum of diseased indicators for water bodies with healthy (open circles) and diseased shrimp populations (solid triangles). Diseased shrimp populations expected in water bodies with a sum of diseased indicators >45 % and a sum of healthy indicators of <15 %, while healthy shrimp populations expected at diseased indicators <45 % and healthy indicators >15 %
Discussion
A widely accepted paradigm on the disease in aquaculture is the result of the unbalanced interactions among the host, environmental variables, and the surrounding microflora, that is, dramatic changes in bacterioplankton community could negatively affect host health (Snieszko 1974; Boutin et al. 2013; Engering et al. 2013). Consistently, we observed a robust dynamics of bacterial assemblages during the monitored period, with especially pronounced separation between PHS and PDS (Fig. 1). In this study, we tested whether indicator phylotypes could be served as independent variables to predict the incidences of shrimp diseases.
Multiple lines of evidence have indicated that aquatic microbial communities are highly temporal dynamics (Gilbert et al. 2009; Or et al. 2012; Teeling et al. 2012; Xiong et al. 2014b). Similarly, we found a modest contribution of OTU-level dynamics over time, reflected by the bacterial communities that appeared to be more cohesive (i.e., H45 vs. H55 and H63 vs. H69) between consecutive sampling points than others (Fig. 1). However, the bacterial communities were significantly distinct (P < 0.05) between PDS and every PHS (at different sampling points), even within the same sampling point (D63 vs. H63, Table 1). These variations could be partially caused by the temporal changes in geochemical variables (Fig. S3), but likely also by the outbreak of shrimp disease. In addition, more phyla are affected by the emergence of shrimp disease than sampling time effects, including the predominant phyla of Bacteroidetes and Alphaproteobacteria (Table 1). Therefore, it is most likely that sudden shifts in the bacterioplankton community result in the shrimp disease outbreak, which is consistent with the notion that microorganisms regulate system processes important for shrimp health (Beardsley et al. 2011). The distinct bacterial assemblages associated with the two types of the health status leading us to speculate that the compositional shifts in microbiota could be predictive for the incidences of shrimp disease.
Disentangling the correlations between shrimp disease and surrounding microflora composition will enable a prediction of the incidence of disease, which is the ultimate goal of such microbiome studies. In accordance with this concept, we developed a method to identify indicator phylotypes and consequently obtained 13 indicator families that discriminate the bacterial communities between PDS and PHS, with a high degree of certainty (Fig. 2). This finding is not unexpected because we can generate reasonable proposes based on the knowledge of the biology and ecology of these indicator families. For example, Flavobacteria are heterotrophs that specialize in the degradation of complex organic matter and offer bioavailable substrates to other taxa (Teeling et al. 2012; Williams et al. 2013). Accordingly, a significant decrease of Flavobacteriaceae populations potentially unbalance the microbial loop (Fig. 2), corresponding with chemical oxygen demand (COD) peaked in PDS (Table S1). In contrast, Rickettsiales are known to be parasitic habitants and can result in severe diseases (Parola and Raoult 2001). Further support for this comes from the notion that the functional potential of microbial community could be predictable from phylogenetic structures (Gilbert et al. 2009; Langille et al. 2013). Thus, an increase in relative abundance of these potential pathogenic Rickettsiales could cause the emergences of shrimp disease. Interestingly, about half of the identified indicators affiliated with uncultured families (Fig. 2), suggesting that more efforts should be paid on the characterization of pathogenic bacteria in shrimp aquaculture. Importantly, the accuracy of application of diseased indicator (DI) phylotypes to predict disease is 100 % (relative high accuracy for prediction health, 75 %, Table 3), which, in turn, indirectly confirmed the reliability of our created model for screening indicator phylotypes. In particular, the inaccurately predicted samples were mainly from the initial stages, such as H35 and H45 (Fig. 3), which may be due to the high dynamics of bacterioplankton community over time.
In particular, the combination of those families could indicate shrimp health status. Specifically, if the sum of the relative abundance of the families that represent healthy indicators is greater than 15 %, we could confidently conclude that shrimp population in those ponds is healthy. By contrast, the sum of the relative abundance of the families that represent diseased indicators is much higher (>45 %) for predicting the occurrence of disease (Fig. 4). One potential explanation of this outcome is that some microbial groups show metabolic flexibility and functional redundancy (Wohl et al. 2004; Allison and Martiny 2008); thus, a slight replacement between healthy and diseased bacterial groups does not sufficiently result in the occurrences of disease. Alternatively, disease emergence is usually a consequence of major shifts in the bacterioplankton assemblages that perturbs the natural balance (Woo and Bruno 2011) and subsequently leads to significant changes in the biology of infection (Walker and Mohan 2009). If this is the case, it is reasonable to require much greater proportion (i.e., >45 %) of potential pathogenic populations to accurately predict the explosive emergence of disease. Overall, in a given pond, the sums of the relative abundance of these indicator phylotypes (bio-indicators) show great promise in predicting its health status.
In conclusion, understanding the relationship between changes in bacterioplankton communities and shrimp health/disease could shed light on the pathogenesis and disease prediction. This study contributes the first attempt to apply bacterial indicators for predicting the increased risk of disease occurrence in aquaculture ecosystem. Importantly, family-level variations in phylotype dynamics provide the highest discrimination. From a practical point, this information would be of great importance for designing family-specific primers, then applying quick and cheap qPCR to quantitatively assay the relative abundances of these indicator families (Fig. 4) for predicting the occurrences of shrimp disease. Together, this study exemplifies that the indicator phylotypes could be served as independent variables to predict the incidence of disease with a high degree of accuracy, although additional works are needed to evaluate the utility of this approach.
References
Allison SD, Martiny JBH (2008) Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci USA 105:11512–11519
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
Beardsley C, Moss S, Malfatti F, Azam F (2011) Quantitative role of shrimp fecal bacteria in organic matter fluxes in a recirculating shrimp aquaculture system. FEMS Microbiol Ecol 77:134–145
Berry D, Schwab C, Milinovich G, Reichert J, Mahfoudh KB, Decher T, Engel M, Hai B, Hainzl E, Heider S, Kenner L, Müller M, Rauch I, Strobl B, Wagner M, Schleper C, Urich T, Loy A (2012) Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J 6:2091–2106
Boutin S, Bernatchez L, Audet C, Derôme N (2013) Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS One 8:e84772
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pel[ntilde]la AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336
Chytrý M, Tichý L, Holt J, Botta-Dukát Z (2002) Determination of diagnostic species with statistical fidelity measures. J Veg Sci 13:79–90
Clarke KR (1993) Non-parametric multivariate analysis of changes in community structure. Aust J Ecol 18:117–143
De Cáceres M, Legendre P (2009) Associations between species and groups of sites: indices and statistical inference. Ecology 90:3566–3574
De Cáceres M, Legendre P, Moretti M (2010) Improving indicator species analysis by combining groups of sites. Oikos 119:1674–1684
Defoirdt T, Sorgeloos P, Bossier P (2011) Alternatives to antibiotics for the control of bacterial disease in aquaculture. Curr Opin Microbiol 14:251–258
Engering A, Hogerwerf L, Slingenbergh J (2013) Pathogen–host–environment interplay and disease emergence. Emerg Microbes Infect 2:e5
Ferreira NC, Bonetti C, Seiffert WQ (2011) Hydrological and water quality indices as management tools in marine shrimp culture. Aquaculture 318:425–433
Fortunato CS, Eiler A, Herfort L, Needoba JA, Peterson T, Crump BC (2013) Determining indicator taxa across spatial and seasonal gradients in the Columbia River coastal margin. ISME J 7:1899–1911
Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T, Somerfield PJ, Huse S, Joint I (2009) Seasonal succession of microbial communities in the Western English Channel using 16S rDNA-tag pyrosequencing. Environ Microbiol 11:3132–3139
Hilbe JM (2009) Logistic regression models. CRC Press, Boca Raton
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clementa JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotech 31:814–821
Lemonnier H, Herbland A, Salery L, Soulard B (2006) “Summer syndrome” in Litopenaeus stylirostris grow out ponds in New Caledonia: zootechnical and environmental factors. Aquaculture 261:1039–1047
Lucas R, Courties C, Herbland A, Goulletquer P, Marteau AL, Lemonnier H (2010) Eutrophication in a tropical pond: Understanding the bacterioplankton and phytoplankton dynamics during a vibriosis outbreak using flow cytometric analyses. Aquaculture 310:112–121
Ma Z, Song X, Wan R, Gao L (2013) A modified water quality index for intensive shrimp ponds of Litopenaeus vannamei. Ecol Indic 24:287–293
McGeogh MA (1998) The selection, testing and application of terrestrial insects as bioindicators. Biol Rev 73:181–201
Or A, Shtrasler L, Gophna U (2012) Fine-scale temporal dynamics of a fragmented lotic microbial ecosystem. Sci Rep 2:207
Parola P, Raoult D (2001) Tick-borne bacterial diseases emerging in Europe. Clin Microbiol Infect 7:80–83
Paerl HW, Dyble J, Moisander PH, Noble RT, Piehler MF, Pinckney JL, Steppe TF, Twomey L, Valdes LM (2003) Microbial indicators of aquatic ecosystem change: current applications to eutrophication studies. FEMS Microbiol Ecol 46:233–246
Snieszko SF (1974) The effect of environmental stress on outbreaks of infectious diseases of fish. J Fish Biol 6:197–208
Sugiura SH, Marchant DD, Wigins T, Ferraris RP (2006) Effluent profile of commercially used low-phosphorus fish feeds. Environ Pollut 140:95–101
Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Jörg Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R (2012) Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611
Walker PJ, Mohan CV (2009) Viral disease emergence in shrimp aquaculture: origins, impact and the effectiveness of health management strategies. Rev Aquaculture 1:125–154
Williams TJ, Wilkins D, Long E, Evans F, DeMaere MZ, Raftery MJ, Cavicchioli R (2013) The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ Microbiol 15:1302–1317
Wohl DL, Arora S, Gladstone JR (2004) Functional redundancy supports biodiversity and ecosystem function in a closed and constant environment. Ecology 85:1534–1540
Woo PT, Bruno DW (2011) Fish diseases and disorders: viral, bacterial and fungal infections (Vol. 3). CABI
Xiong J, Ye X, Wang K, Chen H, Hu C, Zhu J, Zhang D (2014a) The biogeography of the sediment bacterial community responds to a nitrogen pollution gradient in the East China Sea. Appl Environ Microbiol 80:1919–1925
Xiong J, Zhu J, Wang K, Wang X, Ye X, Liu L, Zhao Q, Hou M, Qiuqian L, Zhang D (2014b) The temporal scaling of bacterioplankton composition: high turnover and predictability during shrimp cultivation. Microb Ecol 67:256–264
Zhang D, Wang X, Xiong J, Zhu J, Wang Y, Zhao Q, Chen H, Guo A, Wu J, Dai H (2014) Bacterioplankton assemblages as biological indicators of shrimp health status. Ecol Indic 38:218–224
Zhou J, Fang W, Yang X, Zhou S, Hu L, Li X, Qi X, Su H, Xie L (2012) A nonluminescent and highly virulent Vibrio harveyi strain is associated with “bacterial white tail disease” of Litopenaeus vannamei shrimp. PLoS One 7:e29961
Acknowledgments
This work was financially supported by the National High Technology Research and Development Program of China (863 Program, 2012AA092000), the Experimental Technology Research and Development Project of Ningbo University (SYJS-201405), Natural Science Foundation of Ningbo City (2013A610169) and of Ningbo University (XYL14004), Research Fund from 2011 Center of Modern Marine Aquaculture of East China Sea, and the KC Wong Magna Fund of Ningbo University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jinbo Xiong and Jianlin Zhu contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 579 kb)
Rights and permissions
About this article

Cite this article
Xiong, J., Zhu, J. & Zhang, D. The application of bacterial indicator phylotypes to predict shrimp health status. Appl Microbiol Biotechnol 98, 8291–8299 (2014). https://doi.org/10.1007/s00253-014-5941-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5941-y