
Abstract
The spatial distribution of microbial communities has recently been reliably documented in the form of a distance–similarity decay relationship. In contrast, temporal scaling, the pattern defined by the microbial similarity–time relationships (STRs), has received far less attention. As a result, it is unclear whether the spatial and temporal variations of microbial communities share a similar power law. In this study, we applied the 454 pyrosequencing technique to investigate temporal scaling in patterns of bacterioplankton community dynamics during the process of shrimp culture. Our results showed that the similarities decreased significantly (P = 0.002) with time during the period over which the bacterioplankton community was monitored, with a scaling exponent of w = 0.400. However, the diversities did not change dramatically. The community dynamics followed a gradual process of succession relative to the parent communities, with greater similarities between samples from consecutive sampling points. In particular, the variations of the bacterial communities from different ponds shared similar successional trajectories, suggesting that bacterial temporal dynamics are predictable to a certain extent. Changes in bacterial community structure were significantly correlated with the combination of Chl a, TN, PO4 3-, and the C/N ratio. In this study, we identified predictable patterns in the temporal dynamics of bacterioplankton community structure, demonstrating that the STR of the bacterial community mirrors the spatial distance–similarity decay model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The paradigm that ‘Disease in aquaculture is the result of complex interactions among the host, environmental variables, and the surrounding microflora’ [37] has been supported by a range of studies [25, 27, 49]. These findings indicate the critical role of suitable bacterioplankton for maintaining shrimp health. Specifically, a balance between beneficial and pathogenic bacteria is necessary for successful shrimp cultivation. Although there is evidence that aquatic microbial communities are highly dynamic over time [15, 30, 35, 44], it is still uncertain whether such temporal variations are predictable.
To sustain high production, the feed supply generally exceeds the requirements of cultured shrimp, producing gradual eutrophication and resource variation in the water bodies associated with the culture [13, 29]. This effect may, in turn, trigger the dynamics of microbial communities during shrimp cultivation because resource partitioning is known to be the key contributor to microbial compositional [18, 39] and functional [45, 46] patterns. Several recent studies have consistently shown temporal changes in the microbial communities of shrimp ponds [25, 27, 48]. However, these observational studies focused on specific taxa, such as Vibrio [27, 38], or were based on fingerprint techniques [4] and thus might provide limited and potentially biased information on the community-level response to increasing eutrophication on a temporal scale. In particular, it is unclear whether these dynamics are random or predictable in such artificially manipulated ponds (a constantly changing environment), although discernible temporal patterns have been detected in natural aquatic ecosystems [15, 44].
Spatial patterns in microbial communities have long been regionally characterized in terms of a distance–similarity decay relationship, i.e., communities in sites separated by a greater geographical distance show more dissimilarity [17, 47]. In contrast, patterns in similarity–time relationships (STRs) have received much less attention [35]. Encouragingly, Jones and colleagues [22] have postulated equivalent temporal and spatial scales for the variation found in aquatic bacterial community composition. Thus, we adapted a mathematical equation analogous to that used for the spatial scale: S = cT w [16], where the scaling exponent w is a reflection of the turnover rate of microbial communities. The estimation of accumulative richness over time is very important for understanding the maintenance of microbial diversity in a given ecosystem [42]. Thus, existing temporal scaling studies have generally focused on the taxa–time relationship, representing the accumulative richness over time based on censuses of diverse ecosystems [23, 34, 40, 42, 43]. As a result, it is not clear whether the relationships between time and the similarities among microbial communities mirror those detected for the spatial scale.
Presumably, shrimp ponds are renewed habitats that are sterilized before the introduction of juveniles and identically managed during the entire culture period. For these reasons, they offer an ideal, full-scale setting in which to conduct detailed studies to test microbial temporal dynamics. Individual bacterial taxa differ substantially in their metabolic capabilities, e.g., the difference between specialist and generalist taxa. Thus, changes in the substrate have the potential to shape bacterial community structure [19, 46, 49]. Therefore, we hypothesize that the variations in the bacterial community are correlated with a few environmental variables (driving forces) and that the temporal dynamics of the community may follow the spatial power law [16], as temporal variation and spatial variation are highly comparable [22]. To verify these hypotheses, we collected water samples from shrimp (Litopenaeus vannamei) culture ponds, using amplicon pyrosequencing of 16S rDNA to characterize the temporal dynamics of the planktonic bacterial community and the abiotic factors that drive this pattern and to explore whether the STRs followed the model found for the spatial scale.
Materials and Methods
Experimental Design and Water Sample Collection
The shrimp ponds investigated in this study are located in Zhanqi, Ningbo, eastern China (29°32′N, 121°31′E) in an area measuring 300 by 600 m. These 30 ponds are approximately the same size (2,000 m2) and are identically managed in terms of sea water inputs, daily water exchange rate (5 %) and depth (1.5 m), shrimp stocking density (360,000 ind/pond), feed type and schedule. The ponds are located in greenhouses to maintain a relatively stable temperature during the cool season. Bottom aeration is applied to maintain a suitable level of dissolved oxygen. Shrimp (Litopenaeus vannamei) juveniles were introduced into the ponds on 25 March 2012. The water samples were collected at various time points separated by 6 to 10 days (over a span of 42 days, from 29 April to 10 June) in six different ponds at a depth of 50 cm below the water surface, corresponding to 35, 45, 55, 63, 69 and 77 days after shrimp inoculation. To minor the spatial variability within ponds, samples were chosen from four representative points (similar locations among the ponds) and combined to form a composite biological replicate sample representing a given pond and time point. In total, we collected 36 water samples (six ponds × six time points). The samples were immediately transported (within 4 h) to the laboratory in an icebox.
Water temperature and pH were recorded with appropriate sensors at a depth of 50 cm. The concentrations of total organic carbon (TOC), total nitrogen (TN), total phosphate (TP), NO3 - and PO4 3+ and the chemical oxygen demand (COD) were analyzed following standard methods [2]. For the measurement of chlorophyll a (Chl a), a water sample was filtered through Whatman 25 mm GF/F filters and extracted in 90 % dimethylformamide (N,N-dimethyl formamide) for 24 h at 48 °C. The concentration of Chl a in the supernatant was determined using a spectrophotometer (UV-1601, Shimadzu, Japan).
DNA Extraction
On the sampling days, approximately 1 L of water for DNA extraction was prefiltered through nylon mesh (100-μm pore size). The samples were subsequently filtered onto a 0.2-μm membrane (Millipore, Boston, MA, USA). Community DNA was extracted using a Power Soil® DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) according to the manufacturer’s protocol. The gDNA extracts were quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA) and stored at -80 °C prior to amplification.
Bacterial 16S rRNA Amplification and 454 Sequencing
An aliquot (50 ng) of DNA from each sample was used as the template for amplification. The V1–V3 hypervariable regions of bacterial 16S rRNAs (Escherichia coli positions 27 F–519R) were amplified using the primer set 27 F: AGAGTTTGATCMTGGCTCAG with the Roche 454 ‘A’ pyrosequencing adapter and a unique 10 bp barcode sequence and the primer 519R: GWATTACCGCGGCKGC-TG with the Roche 454 ‘B’ sequencing adapter at the 5′-end of each primer. This region furnished nearly the same resolution as that of the nearly full length sequence [23]. Each sample was amplified in triplicate with a unique barcode primer (in a 50 μl reaction system) under the following conditions: 30 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s, with a final extension at 72 °C for 10 min. Polymerase chain reaction (PCR) products for each sample were combined and purified with a PCR fragment purification kit (TaKaRa Biotech, Japan).
An equimolar amount of PCR products (assuming that amplicons of the same size had a similar molar mass) for each sample were combined in a single tube to be run on a Roche FLX 454 pyrosequencing machine (Roche Diagnostics, Branford, CT, USA), producing reads from the forward direction 27 F with the barcode.
Processing of Pyrosequencing Data
Sequencing reads were demultiplexed, quality filtered and denoised using the Quantitative Insights Into Microbial Ecology (QIIME) workflow [3]. Specifically, the bacterial reads whose length was outside the bounds of 200 and 450 bp and cases in which the homopolymer run exceeds 6 were removed by PyroNoise [9], then sequences with the same barcode were assigned into the same sample [3]. Bacterial phylotypes were identified using uclust [12] and assigned to operational taxonomic units (OTUs, 97 % cutoff). The most abundant sequence from each phytotype was selected as the representative sequence and was aligned using PyNAST [9]. Taxonomic identity of each phylotype was determined using the Greengenes database [10]. To correct for varying sampling efforts, we used a randomly selected subset of 4,500 sequences (corresponding to the smallest sequencing effort for any of the samples) per sample to calculate diversities and distances between samples.
Statistical Analysis
A one-way analysis of variance (ANOVA) was used to test for significant differences in alpha diversity across the sampling points using SPSS 13.0 software. Nonmetric multidimensional scaling (NMDS) and principal coordinates analysis (PCoA) were implemented to evaluate the overall differences in bacterial community structure to determine changes in beta diversity [26]. A nonparametric permutational multivariate analysis of variance (perMANOVA) was conducted to test the effects of various sampling times on microbial community variations, and a similarity percentage analysis (SIMPER) was applied to identify the taxa that were primarily responsible for observed differences among the sampling points based on PAST [5, 20]. To reduce multicollinearity, a canonical correspondence analysis (CCA)-based variance inflation factor (VIF) was calculated to identify the common sets of environmental variables important to bacterial community composition. These analyses were performed in R v.2.11.0 with the ‘vegan’ package [33].
Temporal Turnover Rate Estimation and Taxa–Time Relationship
STRs were determined using a contiguous sampling approach with the power law model S = cT w. Here, the scaling exponent w was considered an index of the temporal turnover rate of the bacterial community, following the idea of spatial turnover [16]. The Sørensen index puts more weight on joint occurrences than on mismatches [36], and thus the contiguous STRs estimate the cumulative decrease in the Sørensen similarities among the microbial communities in a time series framework. Due to the temporal sampling design, the data points within each pond were not independent; we calculated the bacterial community similarity between D35 (the first sampling point) and other sampling points (D45, D55, D63, D69 and D77), and we then averaged the similarity across the sampling period. The power law exponent w was estimated directly with a linear regression based on the function expressed in log–log space: logS = logc + wlogT. A similar model was applied to evaluate the taxa–time relationship, as described elsewhere [16, 40]; please see the detailed information in the legend of Fig. 6.
Results
Physicochemical Characteristics of the Water Samples
The principal physicochemical characteristics of the water samples are summarized in Table S1. The water temperature and pH values were relatively stable, ranging from 27.3 to 30.9 °C due to culturing in the greenhouses and from 6.18 to 7.05, respectively. The environmental parameters, especially NO3 - and PO4 3+, were highly variable in the culture ponds, whereas TOC consistently (r = 0.730, P < 0.001, Pearson correlation) increased over the shrimp harvest cycle. Furthermore, the increase in nutrient (TP and NO3 -) concentrations was paralleled (P < 0.05 in both cases) by a gradual increase in the Chl a concentration. This result is consistent with the findings of previous reports on shrimp culture procedures [27, 48].
Distribution of Taxa and Phylotypes
The sequencing efforts yielded eight samples with low-quality reads, of which four were from D35 and four from D45. Unfortunately, we did not have a sufficient amount of DNA for repeated pyrosequencing. To ensure sufficient sequencing depth, these eight samples were excluded from further analysis. Following this procedure, we obtained a total of 195,640 quality sequences and 4,502–8,285 sequences per sample (mean = 6987) and were able to classify 95.3 % of those sequences across the samples. The dominant phyla or classes across the samples were Actinobacteria (averaged relative abundance, 11.9 %), Alphaproteobacteria (20.5 %), Gammaproteobacteria (6.7 %), Flavobacteria (23.5 %) and Sphingobacteria (6.0 %), representing more than 68 % of the bacterial reads (Fig. 1). Although no consistent pattern (linear increase or decrease) emerged for a dominant phylum or class, several general patterns were observed, e.g., the relative abundance of Alphaproteobacteria and Sphingobacteria decreased, whereas that of Gammaproteobacteria and Betaproteobacteria increased over time (Fig. 1). Strikingly, the taxa that contributed to the overall differences in the bacterial community were primarily affiliated with these dominant groups (Actinobacteria, Alphaproteobacteria and Flavobacteria; Table S2). The alpha diversity including the number of operational taxonomic unit (OTUs; phylotypes) and the Shannon index was relatively stable except for the D35 communities (Fig. S1). In addition, there were no significant correlations between the estimated diversities and either the time of sampling or the environmental variables (r < 0.4, P > 0.5 in all cases).

Relative abundances of the dominant bacterial phyla (relative abundance >1 %) in water samples separated according to sampling time and the average across the samples. Relative abundances are based on the proportional frequencies of those DNA sequences that could be classified at the phylum level, with the exception that the predominant phyla of Bacteroidetes and Proteobacteria were grouped at the class level
Bacterial Community Structure
Based on the detected OTUs across the samples, an NMDS ordination analysis clearly revealed the continuous succession of bacterioplankton assemblages during our monitored shrimp-farm culture, primarily separated by the first axis (Fig. 2a), although the community richness and diversities did not vary dramatically over time (Fig. S1). The linear function showed a significant correlation (Pearson coefficient, ρ = 0.961, P < 0.001) between NMDS axis 1 (as a proxy for the bacterial community dissimilarity) and sampling time (Fig. 2b). The patterns were further corroborated by a dissimilarity test (perMANOVA), which demonstrated that sampling time was an important factor in determining community composition (global F = 1.80, P = 0.001). Note that the community compositions did not vary dramatically between consecutive sampling points, e.g., D45 vs. D55 and D63 vs. D69. However, they changed significantly (P < 0.05) over longer sampling intervals, e.g., D45 vs. D63 and D63 vs. D77 (Table 1). Overall, the results clearly demonstrated the temporal dynamics of the bacterioplankton composition over a short period.

Nonmetric multidimensional scaling (NMDS) plot derived from the Jaccard distances between water samples (a) with symbols coded by sampling time, and the first component from NMDS of the Jaccard distances regressed against sampling time using a linear function for the bacterial community (b)
Linking Bacterial Community Structure to Environmental Conditions
To link the taxonomic structure of the microbial communities with the water properties and Chl a (as a proxy of photosynthetic potential), CCAs of the bacterial communities were performed for each of the ten environmental variables (Table S1). Six of these variables (TOC, TN, PO4 3-, COD, C/N ratio and Chl a) were selected based on P values less than 0.05. These six environmental variables and the bacterial communities were then used in the CCA. The VIFs for TOC and COD were greater than 20, indicating significant correlations between the environmental variables (multicollinearity, data not shown). For this reason, TOC and COD were removed and the CCA repeated. The VIFs for the remaining four variables including TN (P = 0.001), PO4 3+ (P = 0.030), the C/N ratio (P = 0.001) and Chl a (P = 0.007) decreased to less than 2. The corresponding CCA biplot revealed that the variation was significantly (F = 1.32, P = 0.005) correlated with the combination of the variables (Fig. 3). In particular, the bacterial communities in the later periods (D63 to D77) were distinct from those in the early periods (D35 and D45), separated primarily by the first axis, which was positively correlated with the C/N ratio and Chl a and negatively correlated with TN and PO4 3+ (Fig. 3).

Canonical correspondence analysis (CCA) of detected OTUs and the selected water biogeochemistry parameters. The percentage of variation explained by each axis is shown
Similarity–Time and Taxa–Time Relationships
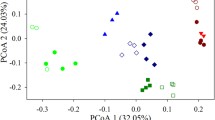
To evaluate the turnover rate of the bacterioplankton community, an STR was applied to assess the temporal dynamics of the overall bacterial assemblage in the artificial shrimp ponds. The slope of the STR was estimated by a linear regression involving the log-transformed similarities of the bacterial communities and the sampling times. A significant (P = 0.002) STR was observed with the exponent w = 0.400 (Fig. 4). Strikingly, the direction of change of the bacterial community appeared to be predictable to a certain extent, i.e., the trajectories of bacterial variation were similar among the ponds during the monitored period (Fig. 5). Consistently, if we retained four time points (D55, D63, D69 and D77) for the six ponds, the ponds appeared to share parallel trajectories (Fig. S2). In particular, there was a significant correlation (P < 0.001) between the cumulative observed OTUs and the days between observations, with a temporal turnover of bacterial taxa of 0.393 (Fig. 6).

Similarity–time relationships (STRs) for bacterial communities. The power-law exponent w was estimated directly with a linear regression (log–log space approach) fit between the average Sørensen similarity values and days between observation in the shrimp ponds

Principal coordinates analysis (PCoA) plots derived from the Jaccard distances between water samples with symbols coded by ponds. To minimize unequal sampling effects (here due to sequencing problems), ponds with six time points (a) and four time points (b) are shown separately. In a given pond, the arrows show the successional direction over time

The taxa–time relationship for bacterial communities. The power law exponent w was estimated directly with a linear regression law (log–log space approach) fit between the cumulative observed OTUs and days between observations in the shrimp ponds. The first observation was at day 35 for ponds 2 and 4 and at day 55 for ponds 1, 3, 5, and 6
Discussion
The initial aim of this study was to characterize the correlation between the bacterial community and the occurrence of shrimp disease. For this reason, we only collected samples covering the periods during which the risk of a disease outbreak was high (58 ± 8 days after the introduction of juvenile shrimp) [24]. Given the high temporal variability of the microbial composition [30], our study appears to have used a relatively coarse sampling frequency. However, it has been shown that the effect of sampling is most likely minor (Fig. 2b) in terms of the estimation of the overall turnover rate [34].
Deterministic Role of Environmental Factors for the Bacterial Community in Shrimp Ponds
Intensive shrimp farming generally produces gradual eutrophication [29], whereas bacterioplankton communities are extremely sensitive and reactive to subtle environmental changes [30]. Consistently, we observed robust dynamics of bacterial assemblages, namely, a decrease in the similarity of the bacterial communities as the length of the census period increased (Fig. 4), accompanied by an unchanging value of diversity (Fig. S1). The temporal changes in the bacterial communities were significantly correlated with the combination of Chl a, TN, PO4 3- and the C/N ratio (Fig. 3), consistent with the finding that substrate availability creates ecological niches in which specialized populations bloom [39]. Likewise, chlorophyll a (Chl a) has been widely applied as a proxy for phytoplankton abundance and biomass [11], which affects the temporal succession of bacterial community composition [31].
Spatial and Temporal Variations of the Microbial Community Share a Similar Power Law
Previous studies have shown that the total cell abundance of bacterioplankton community tends to vary substantially less than the community composition over time [7, 8]. Two basic mechanisms shape population dynamics and function over time, the ‘adjustment scenario’ and the ‘replacement scenario’ [7]. Our results appear consistent with the latter case, in which a continuous change in resources triggers shifts in the bacterioplankton composition involving intrinsically different taxa (Fig. 6). Multiple lines of evidence have indicated that community structure determines the final metabolic response to environmental change including a strong correlation between the rate of change in bacterial community structure and functional capacities [6] and also including the rate of the ecosystem processes affected by microbial composition [1, 46]. Thus, a better understanding of bacterial temporal dynamics is critical to predict ecosystem function, although consistency is difficult to maintain.
The exponent (w = 0.400) of the STRs for the bacterial communities of the shrimp ponds is greater than the slopes (0.02 to 0.03) of the linear relationships observed in bacterial succession in several habitats [35]. One possible explanation for this discrepancy is that we detected much diverse rare species (resulting in a low pairwise similarity) with the pyrosequencing technique, whereas Shade et al. [35] collected data with a much lower sequencing depth, ranging from 486–3526 reads per sample. Note that neither the sequencing depth found by the Shade et al. study nor the sequencing depth found by our study reached saturation (rarefaction curve not shown). It has been proposed that trends in community structure among samples are not particularly sensitive to sequencing depth, e.g., 1,000 denoised sequences per sample explained ~90 % of the variation in beta diversities [28]. Alternatively, it is possible that our slope was overestimated to a certain degree due to stochastic processes [41, 50] such as daily water discharge and influent effects during shrimp culture. Despite this possible influence, the slope of the taxa–time relationships found in our study (Fig. 6) is comparable to the rate of accumulation of bacterial taxa in other habitats [34, 40, 42]. In addition, the taxa primarily contributing to the temporal dynamics of the bacterial community are numerically abundant (Table S2). Furthermore, we found that the bacterial temporal dynamics were significantly correlated with a few environmental parameters (Fig. 3), suggesting that deterministic factors, rather than stochastic processes, were the drivers of temporal variation in the bacterial communities examined in this study.
Predictability of Bacterial Community Dynamics in Shrimp Ponds
It has been shown that the phylogenetic compositions of microbial communities are more similar at adjacent sampling points [15, 34]. Similarly, we found a modest contribution of strain–level variations over time, reflected by certain bacterial communities that appeared to be more cohesive than others (e.g., H45 vs. H55 and H63 vs. H69) between consecutive sampling points (Fig. 2). Additionally, the different ponds sampled on the same day appeared to harbor more similar bacterial communities and followed parallel successional patterns over time (Fig. 5), which clearly indicates that the temporal variability in bacterial community structure in the shrimp ponds was not random but, rather, predictable over the period monitored by our study. Note that communities that were more similar between samples were found at later stages such as D63 vs. D69 and D69 vs. D77 (Table 1 and Fig. 2a). Thus, the rate of change in similarity might decrease at longer time scales. This pattern is consistent with the similarity–distance relationships, i.e., the correlation between community similarity and spatial distance disappeared with steady increases in the geographic distance [17]. Thus, additional studies are required to verify this hypothesis over the entire duration of shrimp culture.
In particular, we presented tangible evidence that the STRs follow the spatial scale model. As has been asserted recently, the changes in environmental characteristics that occur over time are equivalent to aquatic spatial heterogeneity [22], resulting in similar ecological processes driving community dynamics. In fact, there is a long history of substituting space for time to evaluate chronosequences in macroecology [14, 21, 32]. However, this method has been criticized for its assumption of stable biogeochemical factors [21]. We know that such stability cannot be assumed in this case. Accordingly, ecological studies need to shift away from the pattern of cumulative species (with alpha diversity generally unchanged over time) to the examination of community beta diversity in real time as well as that of the underlying ecological processes.
Conclusions
In this study, we provide direct evidence to demonstrate that the temporal dynamics of the similarities (the beta diversity) of the bacterial community, rather than the accumulative richness of taxa, mirrors the distance–similarity decay relationship for microbes [16, 47]. In particular, the successional trajectory is predictable to a certain extent, at least on this short-term (42-day) scale. Overall, we demonstrate that the patterns of the temporal dynamics of bacterial community structure are predictable and that bacterial spatio-temporal variations share a similar power law model.
References
Allison SD, Martiny JBH (2008) Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci U S A 105:11512–11519
APHA (1976) Standard methods for the examination of water and wastewater 14ed. APHA American Public Health Association
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows integration and analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Case M, Leca EE, Leitaoc SN, SantAnna EE, Schwamborn R, De Moraes Junior AT (2008) Plankton community as an indicator of water quality in tropical shrimp culture ponds. Mar Pollut Bull 56:1343–1352
Clarke KR (1993) Non-parametric multivariate analysis of changes in community structure. Aust J Ecol 18:117–143
Comte J, Del Giorgio PA (2010) Linking the patterns of change in composition and functional capacities in bacterioplankton successions along environmental gradients. Ecology 95:1466–1476
Comte J, Del Giorgio PA (2011) Composition influences the pathway but not the outcome of the metabolic response of bacterioplankton to resource shifts. PLoS One 6:e25266
Cotner JB, Biddanda BA (2002) Small players, large role: Microbial influence on biogeochemical processes in pelagic aquatic ecosystems. Ecosystems 5:105–121
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072
DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM et al (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34:394–399
Domingues RB, Barbosa A, Galvão H (2008) Constraints on the use of phytoplankton as a biological quality element within the water framework directive in Portuguese waters. Mar Pollut Bull 56:1389–1395
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Ferreira NC, Bonetti C, Seiffert WQ (2011) Hydrological and water quality indices as management tools in marine shrimp culture. Aquaculture 318:425–433
Fierer N, Nemergut DR, Knight R, Crainej JM (2010) Changes through time: integrating microorganisms into the study of succession. Res Microbiol 161:635–642
Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T et al (2009) The seasonal succession of microbial communities in the Western English Channel using 16S rDNA-tag pyrosequencing. Environ Microbiol 11:3132–3139
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M et al (2004) Spatial scaling of microbial eukaryote diversity. Nature 432:747–750
Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS (2011) The bacterial biogeography of British soils. Environ Microbiol 13:1642–1654
Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD, Brodie EL (2011) Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol 2:1–10
Gómez-Consarnau L, Lindh MV, Gasol JM, Pinhassi J (2012) Structuring of bacterioplankton communities by specific dissolved organic carbon compounds. Environ Microbiol 14:2361–2378
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:1–9
Johnson EA, Miyanishi K (2008) Testing the assumptions of chronosequences in succession. Ecol Lett 11:419–431
Jones SE, Cadkin TA, Newton RJ, McMahon KD (2012) Spatial and temporal scales of aquatic bacterial beta diversity. Front Microbiol 3:318
Kim Y, Jeong J, Wells GF, Park H (2013) General and rare bacterial taxa demonstrating different temporal dynamic patterns in an activated sludge bioreactor. Appl Microbiol Biotechnol 97:1755–1765
Lemonnier H, Herbland A, Salery L, Soulard B (2006) “Summer syndrome” in Litopenaeus stylirostris grow out ponds in New Caledonia: zootechnical and environmental factors. Aquaculture 261:1039–1047
Lemonnier H, Courties C, Mugnier C, Torréton J, Herbland A (2010) Nutrient and microbial dynamics in eutrophying shrimp ponds affected or unaffected by vibriosis. Mar Pollut Bull 60:402–411
Legendre P, Legendre L (1998) Numerical ecology, 2nd English edn. Developments in environmental modeling. Dev Environ Model 20:1–853
Lucas R, Courties C, Herbland A, Goulletquer P, Marteau AL, Lemonnier H (2010) Etrophication in a tropical pond: understanding the bacterioplankton and phytoplankton dynamics during a vibriosis outbreak using flow cytometric analyses. Aquaculture 310:112–121
Lundin D, Severin I, Logue JB, Östman Ö, Andersson AF, Lindström ES (2012) Which sequencing depth is sufficient to describe patterns in bacterial α- and β-diversity? Environ Microbiol Rep 4:367–372
Ma Z, Song X, Wan R, Gao L (2013) A modified water quality index for intensive shrimp ponds of Litopenaeus vannamei. Ecol Indic 24:287–293
Or A, Shtrasler L, Gophna U (2012) Fine-scale temporal dynamics of a fragmented lotic microbial ecosystem. Sci Rep 2:207
Paver SF, Hayek KR, Gano KA, Fagen JR, Brown CT et al (2013) Interactions between specific phytoplankton and bacteria affect lake bacterial community succession. Environ Microbiol 15:2489–2504
Preston FW (1960) Time and space and the variation of species. Ecology 41:612–627
R Development Team (2012) R: A language and environment for statistical computing. http://cran.r-project.org
Redford A, Fierer N (2009) Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb Ecol 58:189–198
Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N (2013) A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7:1493–1506
Sørensen TA (1948) A method of establishing groups of equal amplitude in plant sociology based on similarity of species and its application to analyses of the vegetation on Danish commons. Biol Skr 5:1–34
Snieszko SF (1974) The effect of environmental stress on outbreaks of infectious diseases of fish. J Fish Biol 6:197–208
Sung H, Hsu S, Chen C, Ting Y, Chao W (2001) Relationships between disease outbreak in cultured tiger shrimp (Penaeus monodon) and the composition of Vibrio communities in pond water and shrimp hepatopancreas during cultivation. Aquaculture 192:101–110
Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM et al (2012) Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611
Van Der Gast CJ, Ager D, Lilley AK (2008) Temporal scaling of bacterial taxa is influenced by both stochastic and deterministic ecological factors. Environ Microbiol 10:1411–1418
Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC et al (2013) Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7:1310–1321
Wells GF, Park HD, Eggleston B, Francis CA, Criddle CS (2011) Fine-scale bacterial community dynamics and the taxa–time relationship within a full-scale activated sludge bioreactor. Water Res 45:5476–5488
White EP, Adler PB, Lauenroth WK, Gill RA, Greenberg D et al (2006) A comparison of the species–time relationship across ecosystems and taxonomic groups. Oikos 112:185–195
Wu QL, Hahn MW (2006) High predictability of the seasonal dynamics of a species-like Polynucleobacter population in a freshwater lake. Environ Microbiol 8:1660–1666
Xiong J, Wu L, Tu S, Van Nostrand JD, He Z, Zhou J, Wang G (2010) Microbial communities and functional genes associated with soil arsenic contamination and rhizosphere of the arsenic hyper-accumulating plant Pteris vittata L. Appl Environ Microbiol 76:7277–7284
Xiong J, He Z, Van Nostrand JD, Luo G, Tu S, Zhou J, Wang G (2012) Assessing the microbial community and functional genes in a vertical soil profile with long-term arsenic contamination. PLoS One 7:e50507
Xiong J, Liu Y, Lin X, Zhang H, Zeng J, Hou J et al (2012) Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ Microbiol 14:2457–2466
Xu H, Zhu M, Jiang Y, Al-Rasheid KS (2010) Temporal species distributions of planktonic protist communities in semi-enclosed mariculture waters and responses to environmental stress. Acta Oceanol Sin 29:74–83
Zhang D, Wang X, Xiong J, Zhu J, Wang Y, Zhao Q et al (2013) Bacterioplankton assemblages as biological indicators for shrimp healthy states. Ecol Indic. doi:10.1016/j.ecolind.2013.11.002
Zhou J, Liu W, Deng Y, Jiang Y, Xue K, He s et al (2013) Stochastic assembly leads to alternative communities with distinct functions in a bioreactor microbial community. mBio 4:e00584–12
Acknowledgments
This work was financially supported by the National High Technology Research and Development Program of China (863 Program, 2012AA092000), the Science and Technology Project of the Ministry of Education (Grant No. 208053), the Natural Science Foundation of Ningbo City (2013A610169), the Research Fund from 2011 Center of Modern Marine Aquaculture of East China Sea, and the KC Wong Magna Fund of Ningbo University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiong and Zhu contributed equally to this work
Rights and permissions
About this article
Cite this article
Xiong, J., Zhu, J., Wang, K. et al. The Temporal Scaling of Bacterioplankton Composition: High Turnover and Predictability during Shrimp Cultivation. Microb Ecol 67, 256–264 (2014). https://doi.org/10.1007/s00248-013-0336-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-013-0336-7