
Abstract
An upsurge in the bioeconomy drives the need for engineering microorganisms with increasingly complex phenotypes. Gains in productivity of industrial microbes depend on the development of improved strains. Classical strain improvement programmes for the generation, screening and isolation of such mutant strains have existed for several decades. An alternative to traditional strain improvement methods, genome shuffling, allows the directed evolution of whole organisms via recursive recombination at the genome level. This review deals chiefly with the technical aspects of genome shuffling. It first presents the diversity of organisms and phenotypes typically evolved using this technology and then reviews available sources of genetic diversity and recombination methodologies. Analysis of the literature reveals that genome shuffling has so far been restricted to microorganisms, both prokaryotes and eukaryotes, with an overepresentation of antibiotics- and biofuel-producing microbes. Mutagenesis is the main source of genetic diversity, with few studies adopting alternative strategies. Recombination is usually done by protoplast fusion or sexual recombination, again with few exceptions. For both diversity and recombination, prospective methods that have not yet been used are also presented. Finally, the potential of genome shuffling for gaining insight into the genetic basis of complex phenotypes is also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genome shuffling (GS) is a laboratory evolution method that addresses the limitations of classical strain improvement programmes (SIPs). Classical strain improvement (reviewed by Crook and Alper 2013) is based on high-throughput screening of mutants generated by mutagenesis or laboratory evolution. Best mutants identified in these programs are then used in further rounds of mutagenesis and screening until the desired selected trait is achieved. SIPs are labour intensive and time consuming, typically leading to incremental improvements of 10 % per year, as single mutants are selected and sequentially mutagenized (delCardayre et al. 2013). Pioneered in the early 2000s (Tobin et al. 2001; Zhang et al. 2002), GS consists in the combinatorial evolution of complex phenotypes in whole organisms by genome-scale and recursive recombination of mutants.
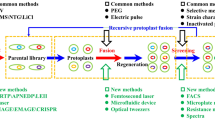
Figure 1 summarizes the general GS workflow, outlining the multiplicity of possible paths. Genetic diversity is introduced in a starting population of interest and recursively recombined to rapidly generate new and potentially beneficial combinations of mutations. Intervening screening or selection steps may be applied at different points in the process to isolate improved mutants, which can be further recombined. This process can be performed repeatedly and stopped whenever the output is deemed satisfactory. Each time a mutant is isolated, it may be submitted to characterization. As will be discussed in this review, system-level characterization of evolved strains can now be used to reveal the complex relationship between the genotype and phenotype of these artificially evolved strains.

Schematic of the general genome shuffling workflow
The patent literature on GS puts strong emphasis on the evolution of microorganisms, but provides examples of methods and applications in plants, fish and other animals (delCardayre et al. 2013). The shuffling of multicellular eukaryotes represents a unique challenge, intensive in time and resources. It would rely on the sexual reproduction cycles of these organisms for recombination and is reminiscent of both existing GS studies in yeast and classical artificial selection schemes (delCardayre et al. 2013). All GS studies published to date are concerned with the improvement of microbial phenotypes (Table 1). This can be attributed to the ease of manipulation, fast growth and inexpensive culturing of microorganisms. The industrial importance of microbes in the production of commodity and specialty chemicals also accounts for their being the focus of GS studies.
The emphasis of this review is on the experimental design of GS experiments to provide readers with directions on how to build their own evolution experiments. It does so by looking at published studies, underlining dominant trends and discussing less common, innovative examples. Table 1 is provided as a guide, listing all the GS studies that, to our knowledge, have been published to date. Examining Table 1, the reader will notice that a little more than 10 years after the first genome shuffling reports, the number of studies in the field has exploded, with the last 4 years providing the largest publication harvests. However, limiting the discussion of GS to examples from the published scientific literature would give an excessively narrow picture of the potential of evolutionary engineering by GS. Patents and patent applications for GS give a much broader view of the method and are a testimony to the scientific and technical possibilities of this technology (see delCardayre et al. 2013 for the most recent patent application). To provide a forward-looking vision of this technology, this review also covers projected yet never applied approaches to GS experiments, hoping it will nourish the imagination of readers in the design of their own experiments.
Steps of the GS process each constitute a section of this review. The first section deals with the species and phenotypes that have been evolved by GS. The second section is dedicated to methods for acquiring diversity in GS experiments. The power of GS relies on the recursive recombination of this genetic diversity, and methods for achieving this are discussed in the third section. Critical to the strain evolution process is the choice of an appropriate screening or selection method. Whether desirable mutants within a combinatorial library can be identified and isolated largely dictates the success of GS experiments, but the reader is referred to other publications for a review of recent screening methods (Crook and Alper 2013; Link et al. 2007). As mentioned earlier, organisms generated by GS may provide insight on the genetics behind complex and industrially relevant phenotypes. System-level analysis of GS-evolved strains can be used for further rational engineering, and the last section of this review deals with this promising yet still largely untapped source of information.
Organisms and phenotypes evolved
Enhancements in the production of small molecules are the main objectives of published GS studies. In particular, Streptomyces species producing antibiotics and other molecules have often been evolved by GS (Table 1). Improvement in chemical productivity in a variety of other microbes is also reported (Table 1). Applying GS to improve ethanol titres from Saccharomyces cerevisiae, the workhorse of the fuel and beverage alcohol industries, is also a common objective (Table 1). Improvement in the ability to ferment xylose (Demeke et al. 2013) or co-ferment glucose and xylose (Jingping et al. 2012) was also evolved by GS in recombinant S. cerevisiae. Since S. cerevisiae does not natively ferment xylose, those studies represent attempts at evolving a rationally engineered but suboptimal xylose fermentation phenotype. They highlight the potential of GS at evolving the complex genetic changes that are often required to optimize engineered organisms. In related studies, the pentose-fermenting yeast Scheffersomyces stipitis was also evolved by GS, in one case in conjunction with S. cerevisiae (Bajwa et al. 2010; Zhang and Geng 2012). Aside from its industrial relevance, the ease with which S. cerevisiae and other yeast can undergo sexual recombination contributes to their popularity as GS organisms (discussed below). Improved production of organic acids is another common aim of GS (Table 1), as is the production of proteins and enzymes (Cheng et al. 2009; El-Bondkly 2012; Xu et al. 2011). Similarly, Aspergillus niger was genome shuffled to enhance its capacity to perform transglycosylation reactions for the production of isomaltooligosaccharides (Li et al. 2013b). While the capacity to perform a reaction rather than production titres was the primary aim of the study, the ultimate output remained increased production of industrially relevant molecules.
The second most important phenotype evolved by GS is resistance to chemical-induced stresses such as salt (Cao et al. 2012), acid (Patnaik et al. 2002) or toxic industrial by-products (Pinel et al. 2011). Resistance phenotypes are often linked to production titres: increased resistance to a given compound produced by a microbe often leads to a concomitant increase in its production. In other cases, production conditions themselves are stressful, and improving an organism's resistance to those key stresses also leads to elevated chemical production (Li et al. 2011, 2012; Wei et al. 2008; Zheng et al. 2011a, b, 2012).
Production and resistance phenotypes account for the vast majority of GS studies. Other interesting phenotypes have been reported and are worth mentioning. For example, the bacterium Sphingobium chlorophenolicum, known for its ability to mineralize pentachlorophenol (PCP), was genome shuffled for increased resistance to this toxic pesticide, leading to a parallel increase in degradation capacity (Dai and Copley 2004). A degradative phenotype was similarly evolved in Stenotrophomonas maltophilia for the bioremediation of trinitrotoluene (Lee et al. 2009). In a study related to the antibiotics-producing properties of Streptomyces melanosporofaciens, antagonism for potato pathogens was enhanced by screening for increased inhibition of bacterial growth (Clermont et al. 2011). Data suggested that increased antibiotics production was not the primary cause for the improvement in bactericidal properties of S. melanosporofaciens.
Source of genetic diversity
The first step in any GS experiment is the creation or acquisition of a genetically diverse population to be used for breeding. This section reviews how this diversity can be obtained. Here, diversity is defined as genome-level sequence diversity. Accordingly, the size of the diversity is defined as the number of unique genome sequences. Most studies artificially induce diversity in an otherwise homogeneous starting population. It is also possible to exploit diversity that is naturally available, and the several methods for tapping natural genetic variations for genome shuffling will also be discussed. Figure 2a schematizes the sources of diversity that can be used in GS studies.

a Sources of diversity and b recombination methods for genome shuffling
How does the source and size of genetic diversity in the starting population affect the success of GS? The size of the diversity in the initial population depends on its source: methods of mutagenesis that induce point mutations may generate pools as large as the target genome, while focused libraries (discussed below) will be smaller by definition. As the evolutionary engineering process progresses, the diversity profile of the evolved population changes accordingly. Recombination (discussed in a separate section) generates new permutations, adding a layer of complexity to the diversity landscape while intervening selections steps weed out mutations. The latter can have profound effects on the evolutionary process. Stringent and frequent selection may eliminate mutations that, if properly recombined with other mutations, could display beneficial epistatic interactions. It is therefore generally advisable to select permissively at the beginning of a GS experiment, increasing stringency as improved mutants are isolated. An excessively small pool may also result from stringent selection, potentially leading to a hasty plateau in improvement. On the contrary, a selection pressure that is too permissive may slow down evolution by allowing neutral or deleterious mutations to clutter the pool. An example is the evolution of S. cerevisiae for improved tolerance to pulping effluents. Two selection schemes were performed in parallel: in the first, only mutants superior to wild type were selected each round, while the second did not discard unimproved mutants. The result was a considerably faster evolution in the stringently selected pool (Pinel et al. 2011).
Mutagenesis
Chemical or physical mutagens are most commonly used to induce genetic diversity in GS experiments (Table 1). It is generally assumed that random mutagenesis unbiasly covers the entire genome (Crook and Alper 2013), in spite of evidence that nuance this assumption (Wang et al. 1991). Moreover, it does not require any prior knowledge on the genetic basis of the phenotype of interest. Frequently used chemical mutagens include nitrosoguanidine (NTG) and ethylmethylsulfonate (EMS), while ultraviolet light is widely used as an ionizing radiation to alter the genetic material of microbes. The reader is referred to other reviews and method articles for details on the use of these and other common mutagens (Crook and Alper 2013; Hopwood 1970; Spencer and Spencer 1996; Winston 2008). Mutagens are often used in combination. Although facultative, this is done to increase the diversity of induced mutations, as each mutagen has specific mechanisms of action that lead to different mutational specificities (Cupples and Miller 1989; Setlow 1966; Singer 1981) that vary between species (Hampsey 1991). Mutator strains represent another approach to mutagenesis. Mutator phenotypes stem from various defects (DNA proofreading, mutator tRNAs, etc.) that increase mutational frequency in affected cells, and much like mutagens, they lead to different mutational spectra (Miller 1996). Although no GS studies have used such strains, they were successful in identifying novel mutants in other studies (Mao et al. 1997; Muteeb and Sen 2010). Mutator strains could be applied to GS, where conditional expression of the mutator phenotype would be an attractive means of controlling mutagenesis. Finally, transposon mutagenesis is another method for inducing diversity, which may benefit applications for which whole-gene knockouts are sought (Hamer et al. 2001).
Prior knowledge on the genetic determinants of the phenotype of interest enables a more targeted approach to GS. For example, mutagenesis can be limited to a subset of genes and loci deemed critical to the phenotype. Targeted mutagenesis can be achieved using focused libraries of randomly mutated DNA fragments with high identity to target loci. A straightforward approach would be to have DNA fragments chemically synthesized, for example in the form of relatively short oligonucleotides, in which case the directed evolution approach would be highly reminiscent of multiplex automated genome engineering (MAGE) (Wang et al. 2009) and other similar methods (Pirakitikulr et al. 2010). An alternative method would be to amplify loci of interest from genomic DNA (gDNA) and mutate them at random by error-prone PCR, as in global transcription machinery engineering (Lanza and Alper 2012). Means to deliver focused libraries into cells and effect recombination are discussed in a dedicated section below.
Natural diversity
Desired phenotypes can sometime be found in nature, but not in the desired organism, while genes associated to a phenotype of interest may have natural homologs that can be exploited to accelerate the strain evolution process. Only a few examples report the use of natural genetic diversity as a starting point in evolving a strain by GS. In one example, nitrous acid mutagenesis was coupled to interspecies crosses to yield an organism with enhanced lactic acid production from starch (John et al. 2008). An acid-tolerant mutant of Lactobacillus delbrueckii was crossed by protoplast fusion with Bacillus amyloliquefaciens, a bacterium notable for its efficient starch utilization. In this example, the phenotypes sought were found in two distinct organisms, whose genomes were used as starting diversity. Natural diversity was similarly exploited in conjunction with mutagenesis to evolve higher production of ε-polylysine in five species of Streptomyces. In this study, the five species were separately evolved using ultraviolet (UV) and NTG mutagenesis as the source of diversity. Best isolates from all five species were subsequently submitted to interspecies hybridization, exploiting natural diversity to further improve productivity (Li et al. 2013a).
Clermont et al. (2011) used the diversity that naturally exists between two strains of S. melanosporofaciens and one strain of Streptomyces hygroscopicus and fused them to evolve an organism capable of controlling the proliferation of potato pathogens. In another example, Zheng et al. (2011a) compared 15 strains of S. cerevisiae for their resistance to multiple stresses and their ability to produce ethanol. Among those, two superior strains were identified and submitted to recursive mating to generate an enhanced hybrid. In yet another example of exploiting natural diversity for GS, a strain of yeast was evolved to co-ferment glucose and xylose by transforming S. cerevisiae with a whole gDNA preparation from S. stipitis (Zhang and Geng 2012). Isolates from this transformation were further shuffled by retransforming with S. cerevisiae gDNA.
Genome-scale recombination
The choice of recombination method depends on several considerations. The organism will determine whether protoplast fusion, sexual recombination or other methods are feasible. For example, as will be discussed below, sexual recombination is only possible in organisms with characterized mating cycles. Other recombination methods are tightly linked to the source of diversity. DNA fragment libraries, for example, cannot be delivered into cells by protoplast fusion. Protoplast fusion and sexual recombination account for nearly all published studies, while several other genome-scale recombination methods are suited for GS. The recombination methods discussed in this section are illustrated in Fig. 2b.
Protoplast fusion
Protoplast fusion is the most common recombination method in GS and allows recombination between virtually any two or more cells. Protoplasts are cells stripped of their cell wall by digestion with lysosyme, zymolyase or other cell wall-digesting enzymes depending on the type of microorganism. Fusion is promoted by submitting protoplasts to an electric pulse or by incubating them in the presence of PEG or surfactants that alter membrane fluidity. Recombination can then take place with genetic material from two or more cells enclosed within a single plasma membrane. Fusants are thereafter allowed to regenerate, and viable recombinants can be submitted to screening and selection. A common yet facultative step is protoplast inactivation. In this variation, protoplasts are rendered non-viable by exposure to UV light or heat. The only way for protoplasts to recover is to undergo fusion and recombination to repair fatal lesions. This approach prevents cells from the parent population from dominating the recombinant pool, as it results in failure of unfused protoplast to regenerate (Fodor et al. 1978; Zhao et al. 2008). It may also induce further diversity via the action of an inactivating mutagen.
An important advantage of protoplast fusion is that it enables poolwise recombination. In other words, any number of protoplasts can theoretically merge into a single fusant. This was demonstrated in Streptomyces coelicolor where a single round of protoplast fusion was sufficient to combine four different auxotrophic markers into one cell, albeit with low efficiency (Hopwood and Wright 1978). This means that recombination between several mutants can occur at once, potentially accelerating the evolution process by creating more combinations and permutations. Four rounds of recursive fusion were later shown to increase the proportion of recombinants: cells carrying two markers went from 8.4 % of the population after one round to 60 % after four, while those carrying three and four markers increased from 0.73 to 17 % and from 0.00005 to 2.5 %, respectively (Zhang et al. 2002). Protoplast fusion is less efficient in gram-negative than gram-positive bacteria. The periplasm and outer membrane of gram-negative bacteria harbour many important functions that are stripped away during protoplasting, making regeneration more challenging. In E. coli, the highest reported proportion of prototrophs from the fusion of two complementary auxotrophic populations is 0.7 % (Dai et al. 2005). A small number of GS studies in gram-negative bacteria have been published (Dai and Copley 2004; Gong et al. 2007; Lee et al. 2009; Wang et al. 2012a; Zheng et al. 2013b).
Sexual recombination
Sexual recombination is typically used when genome shuffling S. cerevisiae and other yeast (Table 1). This approach takes advantage of the well-characterized mating and sporulation cycles of yeast species to avoid some of the disadvantages of protoplast fusion. Using the natural ability of haploid yeast cells to fuse with one another circumvents the delicate task of generating protoplasts. The strategy also takes advantage of the molecular meiotic machinery for recombination. Using any pair of drug resistance markers, cells that have not mated can be eliminated by selecting for double resistance (Zheng et al. 2011b). Yet, sexual recombination has a number of disadvantages. The most obvious is that it is limited to the subset of organisms with an easily manipulated sexual cycle. A second objection is that it only allows pairwise recombination, whereas other methods enable poolwise recombination. Mating takes place between two cells, allowing for only two genomes to recombine at once. In theory, GS based on sexual recombination could thus require more cycles than protoplast fusion to combine the same set of beneficial mutations into a single cell. However, using auxotrophic strains carrying four different auxotrophic markers, it was possible to generate 35 % double auxotrophs after one round of mating (Pinel et al. 2011), a proportion considerably above what has been reported for protoplast fusion (Zhang et al. 2002). Prototrophs (i.e. individuals that recombined four markers into one cell) represented 0.024 % of the population after two rounds and 0.84 % after three (Pinel et al. 2011). The latter efficiencies are inferior to similar reports for protoplast fusion of S. coelicolor (see above). The same experiment in S. stipitis achieved a proportion of mated cells of 0.05 % (Bajwa et al. 2010), demonstrating the interspecies variability of sexual recombination.
Others recombination methods
Other recombination methods can be envisioned in microbes, many of which will show higher efficiency in gram-negative bacteria. Mechanisms for horizontal gene transfer can be exploited to foster recombination between mutants. For example, in bacterial species with characterized fertility factors, conjugation may be an attractive way of effecting exchanges of genetic information. Phage-mediated transduction and direct transformation may also be exploited to deliver DNA libraries into cells. In their evolution of xylose fermentation in baker's yeast, Zhang and Geng (2012) used existing transformation protocols to deliver entire gDNA preparations from S. stipitis and S. cerevisiae into baker's yeast. This approach has the advantage of being simple and straightforward and is especially attractive for shuffling S. cerevisiae with related species because of the efficient homologous recombination capabilities of this organism.
In organisms lacking well-established transformation protocols, liposomes can be used to deliver DNA into protoplasts. The form in which DNA is delivered can also vary. It may be transformed as pure DNA fragments and left to recombine freely into cells. Recombination efficiency can be promoted by coating DNA with RecA protein prior to transformation (Radding 1989; Révet et al. 1993; Sena and Zarling 1993) or by using a recombination system that uses clustered regularly interspaced short palindromic repeats (CRISPR) (Cong et al. 2013). To promote recombination, the stability of the transformed DNA can be increased by the use of vectors. Suicide vectors (i.e. lacking a functional origin of replication in the recipient host) can be used to insert sequences into cells for recombination while ensuring clearing of the foreign DNA. Replicating vectors and artificial chromosomes can also be used, but will require means of negative selection curing them from the recipient. Recombination before curing is necessary in this approach and may have to be induced or selected. Yeast artificial chromosomes, for example, will recombine during meiosis. In all approaches relying on transformation or liposome delivery rather than cell fusion, an elegant way to perform recursive recombination is the split pool approach (delCardayre et al. 2013). After the initial recombination, the pool of recombinants is split in two. Genetic material is extracted from one pool and delivered into the second pool for recombination.
Bridging the gap between genotype and phenotype
Little attention has been given to the genetic basis of improved phenotypes in GS-evolved strains. Yet, these strains are an opportunity to gain insight into the genetic basis of often poorly understood but industrially important phenotypes. Investigating the genetic changes in GS mutants can therefore prove rewarding for future rational approaches, such as inverse metabolic engineering. Linking phenotype to genotype can also prove interesting from a basic science point of view, contributing to our understanding of the systems biology of complex traits. A relatively few examples of such investigations can be found in the literature.
Examination of changes in gene expression profiles of GS-evolved strains is a powerful means of uncovering the causes of phenotypic improvements. After GS of S. cerevisiae for increased performance in very high gravity (VHG) fermentation, Tao et al. (2012) monitored changes in expression of genes involved in trehalose metabolism. Trehalose is an oligosaccharide tightly associated with stress response in yeast, and analysis of cells revealed the evolved strain accumulated more trehalose. Activity of trehalose-producing enzymes was also augmented, while trehalose degradation activity was decreased. Reverse transcriptase quantitative PCR (RT-qPCR) of genes involved in trehalose metabolism revealed a constitutive expression pattern in the GS-evolved strain, whereas induced expression was observed in the parent. Pulse-field gel electrophoresis revealed chromosomal rearrangements hypothesized to cause the changes in gene expression.
In another example, RT-qPCR was used to probe levels of surfactin synthetase (SrfA) gene expression in B. amyloliquefaciens evolved by GS (Zhao et al. 2012). The shuffled strain, which produced more than 10-fold the surfactin titres of its parent, also contained around 15 times more SrfA mRNA. Also, using qPCR, Jin et al. (2012) investigated gene expression variations in a previously identified GS mutant of Streptomyces pristinaespiralis with increased pristinamycin production (Xu et al. 2008), concentrating on genes known to be involved in pristinamycin synthesis. The expression of two of these genes (snbA, snaB), involved in distinct sections of the synthesis process, declined during prolonged fermentation in the parent strain, while it was maintained in the shuffled mutant. A third gene involved in resistance to the antibiotic was expressed earlier during fermentation by the mutant than by the parent. Restriction fragment length polymorphisms (RFLP) analysis was also used to visualize chromosomal alterations potentially involved in pristinamycin yield improvements. Cloning of fragments present in the mutants but not in the parent strain identified two novel genes hypothesized to play a role in pristinamycin synthesis by S. pristinaespiralis.
A yeast species with potential for flavour enhancement of soy sauce, Zygosaccharomyces rouxii was genome shuffled to yield a strain with increased resistance to high salt concentrations (Cao et al. 2010). In a follow-up study, the causes for this increased resistance were investigated (Wei et al. 2013). The HOG1 mitogen-activated protein kinase is known to activate genes involved in glycerol synthesis in S. cerevisiae, and it was reasoned that the Z. rouxii homolog (ZrHOG1) was a likely hit in the shuffled strain. Sequence comparison of the parental and mutant ZrHOG1 genes revealed two nucleotide substitutions in the open reading frame, resulting in a single amino acid change and a single base change upstream of the start codon. While the amino acid change in ZrHOG1 suggested no obvious changes in protein function, estimation of transcription levels by qPCR pointed to elevated activity in the shuffled mutant. Furthermore, expression of mutant ZrHOG1 in S. cerevisiae led to increased glycerol contents.
The characterization studies reviewed above primarily rely on prior knowledge of the phenotype of interest and target specific genes. More open-ended approaches increase the probability of identifying novel genes and pathways not previously associated with the phenotype of interest. Such approaches may also provide a wider, system-level picture of the changes operated in evolved strains. In one study, a shuffling mutant of Propionibacterium shermanii with improved vitamin B12 production was submitted to a cursory proteomics analysis by 2D-gel electrophoresis. Comparison of the gel profiles of the parent and enhanced strains identified 38 proteins with altered levels, including several enzymes involved directly or indirectly in vitamin B12 synthesis.
In recent years, the cost of whole-genome sequencing has decreased considerably. This enables the detailed investigation of genetic and transcriptional changes in genome-shuffled recombinants. The genome of a previously isolated shuffling mutant of S. cerevisiae with increased resistance to toxic pulping effluents (Pinel et al. 2011) was fully sequenced to identify single-nucleotide polymorphisms (SNPs) responsible for the enhanced phenotype (manuscript submitted). Sequencing identified many SNPs in genes that encode stress-response proteins as well as hitherto unexpected hits.
In a recent study, strains of S. cerevisiae evolved for increased stress resistance and ethanol titres in VHG fermentation were characterized by a combination of physico-chemical and genetic methods that include karyotyping, qPCR, array comparative genome hybridization (aCGH) and RNA sequencing (RNA-Seq) (Zheng et al. 2013a). The report is unique among GS studies, in that it used a chemical mutagen, methyl benzimidazole-2-yl-carbamate (MBC), that mainly induces large-scale structural rearrangements of the genome rather than point mutations. Pulse-field gel electrophoresis showed the evolved strains displayed altered karyotypes compared to parent. qPCR also showed copy number variations throughout the genomes of the mutants. aCGH of the most productive shuffled mutant identified the largest copy number variations on chromosomes 8, 11 and 14. RNA-Seq analysis confirmed the presence of several differentially expressed genes from those chromosomes. Mitotic cell cycle, small-molecule metabolism and stress response were the main functional annotations among differentially expressed genes. For example, catalase and trehalose metabolism genes showed increased transcription. This observation correlated with increased catalase and trehalose titres in all mutants tested. Two other genes with suspected roles in stress resistance (YFL052W and SKN7) had increased transcription in the most productive mutant. Their effect on the stress resistance phenotype was confirmed when overexpressed in the parent background. Together, these results showed a clear link between copy number variations, transcription levels and stress resistance phenotype.
An important challenge of whole-genome sequencing approaches is assessing the contribution of each SNP to the phenotype of interest. Because of synergistic effects and hitchhiker mutations, studying SNPs in isolation may not reveal the importance of each mutation. In addition, relatively small pools of mutations result in large numbers of combinations of two or more mutations to test. Backcrossing GS recombinants with the wild type may eliminate hitchhikers. Focused libraries and other targeted genome engineering approaches based on the sequencing results may also allow identification of critical genes and mutations while potentially affording further phenotypic improvements.
Conclusion
Genome shuffling is a powerful method for the directed evolution of whole organisms and complex phenotypes. It requires a source of genetic diversity, natural or induced, a method for recursive mutant recombination and a robust screen or selection for mutant isolation. We have seen how GS studies have addressed these central requirements and have suggested possible approaches for future studies. To date, applications of genome shuffling have been relatively narrow in scope and method, and it can only be hoped that in the coming years, this technology will find wider application in innovative studies. Furthermore, we have barely started exploring what genome-shuffled mutants can teach us about the systems biology of industrially relevant organisms. The many existing genome-shuffled strains are all potential targets for such investigations by sequencing, transcriptional analysis, proteomics and other methods. The combination of directed evolution and mutant characterization therefore holds promises for both increases in productivity and advancement of basic science.
References
Bajwa PK, Pinel D, Martin VJ, Trevors JT, Lee H (2010) Strain improvement of the pentose-fermenting yeast Pichia stipitis by genome shuffling. J Microbiol Methods 81:179–186
Cao X, Hou L, Lu M, Wang C, Zeng B (2010) Genome shuffling of Zygosaccharomyces rouxii to accelerate and enhance the flavour formation of soy sauce. J Sci Food Agric 90:281–285
Cao X, Song Q, Wang C, Hou L (2012) Genome shuffling of Hansenula anomala to improve flavour formation of soy sauce. World J Microbiol Biotechnol 28:1857–62
Chen X, Wei P, Fan L, Yang D, Zhu X, Shen W, Xu Z, Cen P (2009) Generation of high-yield rapamycin-producing strains through protoplasts-related techniques. Appl Microbiol Biotechnol 83:507–512
Cheng Y, Song X, Qin Y, Qu Y (2009) Genome shuffling improves production of cellulase by Penicillium decumbens JU-A10. J Appl Microbiol 107:1837–1846
Clermont N, Lerat S, Beaulieu C (2011) Genome shuffling enhances biocontrol abilities of Streptomyces strains against two potato pathogens. J Appl Microbiol 111:671–682
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–23
Crook N, Alper HS (2013) Classical strain improvement. In: Patnaik R (ed) Engineering complex phenotypes in industrial strains. Wiley, Hoboken, pp 1–33
Cupples CG, Miller JH (1989) A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A 86:5345–9
Dai M, Copley SDC-P (2004) Genome shuffling improves degradation of the anthropogenic pesticide pentachlorophenol by Sphingobium chlorophenolicum ATCC 39723. Appl Env Microbiol 70:2391–2397
Dai M, Ziesman S, Copley SD (2005) Visualization of protoplast fusion and quantitation of recombination in fused protoplasts of auxotrophic strains of Escherichia coli. Metab Eng 7:45–52
delCardayre S, Tobin MB, Stemmer WPC, Ness JE, Minshull JS, Patten PA, Subramanian VM, Castle LA, Krebber CM, Bass SH, Zhang Y-X, Cox AR, Huisman GW, Yuan L, Affholter J (2013) Evolution of whole cells and organisms by recursive sequence recombination. USPTO 8377681B2.
Demeke MM, Dietz H, Li Y, Foulquié-Moreno MR, Mutturi S, Deprez S, Den Abt T, Bonini BM, Liden G, Dumortier F, Verplaetse A, Boles E, Thevelein JM (2013) Development of a d-xylose fermenting and inhibitor tolerant industrial Saccharomyces cerevisiae strain with high performance in lignocellulose hydrolysates using metabolic and evolutionary engineering. Biotechnol Biofuels 6:89
El-Bondkly AMA (2012) Molecular identification using ITS sequences and genome shuffling to improve 2-deoxyglucose tolerance and xylanase activity of marine-derived fungus, Aspergillus sp. NRCF5. Appl Biochem Biotechnol 167:2160–73
El-Gendy MM, El-Bondkly AM (2011) Genome shuffling of marine derived bacterium Nocardia sp. ALAA 2000 for improved ayamycin production. Antonie Van Leeuwenhoek 99:773–780
Fodor K, Demiri E, Alföldi L (1978) Polyethylene glycol-induced fusion of heat-inactivated and living protoplasts of Bacillus megaterium. J Bacteriol 135:68–70
Gao X, Zhao H, Zhang G, He K, Jin Y (2012) Genome shuffling of Clostridium acetobutylicum CICC 8012 for improved production of acetone-butanol-ethanol (ABE). Curr Microbiol 65:128–32
Gong GL, Sun X, Liu XL, Hu W, Cao WR, Liu H, Liu WF, Li YZ (2007) Mutation and a high-throughput screening method for improving the production of epothilones of Sorangium. J Ind Microbiol Biotechnol 34:615–623
Guan N, Liu L, Zhuge X, Xu Q, Li J, Du G, Chen J (2012) Genome shuffling improves acid tolerance of Propionibacterium acidipropionici and propionic acid production. In: Taylor JC (ed) Advances in chemistry research, vol 15. Nova, New York, pp 143–152
Hamer L, DeZwaan TM, Motenegro-Chamoro MV, Frank SA, Hamer JE (2001) Recent advances in large-scale transposon mutagenesis. Curr Opin Chem Biol 5:67–73
Hampsey M (1991) A tester system for detecting each of the six base-pair substitutions in Saccharomyces cerevisiae by selecting for an essential cysteine in iso-1-cytochrome-c. Genetics 128(1):59–67
Hida H, Yamada T, Yamada Y (2007) Genome shuffling of Streptomyces sp. U121 for improved production of hydroxycitric acid. Appl Microbiol Biotechnol 73:1387–1393
Hopwood DA (1970) The isolation of mutants. In: Norris JR, Ribbons DW (eds) Methods in microbiology, vol 3A. Academic, London, pp 363–433
Hopwood DA, Wright HM (1978) Bacterial protoplast fusion: recombination in fused protoplasts of Streptomyces coelicolor. Mol Gen Genet 162:307–317
Hou L (2010) Improved production of ethanol by novel genome shuffling in Saccharomyces cerevisiae. Appl Biochem Biotechnol 160:1084–1093
Jin ZH, Xu B, Lin SZ, Jin QC, Cen PL (2009) Enhanced production of spinosad in Saccharopolyspora spinosa by genome shuffling. Appl Biochem Biotechnol 159:655–663
Jin Q, Jin Z, Zhang L, Yao S, Li F (2012) Probing the molecular mechanisms for pristinamycin yield enhancement in Streptomyces pristinaespiralis. Curr Microbiol 65:792–8
Jingping G, Hongbing S, Gang S, Hongzhi L, Wenxiang P (2012) A genome shuffling-generated Saccharomyces cerevisiae isolate that ferments xylose and glucose to produce high levels of ethanol. J Ind Microbiol Biotechnol 39:777–87
John RP, Gangadharan D, Madhavan Nampoothiri K (2008) Genome shuffling of Lactobacillus delbrueckii mutant and Bacillus amyloliquefaciens through protoplasmic fusion for L-lactic acid production from starchy wastes. Bioresour Technol 99:8008–15
Lanza A, Alper H (2012) Using transcription machinery engineering to elicit complex cellular phenotypes. Methods Mol Biol 813:229–248
Lee BU, Cho YS, Park SC, Oh KH (2009) Enhanced degradation of TNT by genome-shuffled Stenotrophomonas maltophilia OK-5. Curr Microbiol 59:346–351
Li WFF, Ji J, Wang G, Wang HYY, Niu BLL, Josine TLL (2011) Oxidative stress-resistance assay for screening yeast strains overproducing heterologous proteins. Genetika 47:1175–1183
Li S, Li F, Chen X-S, Wang L, Xu J, Tang L, Mao Z-G (2012) Genome shuffling enhanced ε-poly-L-lysine production by improving glucose tolerance of Streptomyces graminearus. Appl Biochem Biotechnol 166:414–23
Li S, Chen X, Dong C, Zhao F, Tang L, Mao Z (2013a) Combining genome shuffling and interspecific hybridization among Streptomyces improved ε-poly-l-lysine production. Appl Biochem Biotechnol 169:338–50
Li W, Chen G, Gu L, Zeng W, Liang Z (2013b) Genome shuffling of Aspergillus niger for improving transglycosylation activity. Appl Biochem Biotechnol 172(1):50–61
Link AJ, Jeong KJ, Georgiou G (2007) Beyond toothpicks: new methods for isolating mutant bacteria. Nat Rev Microbiol 5:680–8
Liu J-J, Ding W-T, Zhang G-C, Wang J-Y (2011) Improving ethanol fermentation performance of Saccharomyces cerevisiae in very high-gravity fermentation through chemical mutagenesis and meiotic recombination. Appl Microbiol Biotechnol 91:1239–46
Luo J-M, Li J-S, Liu D, Liu F, Wang Y-T, Song X-R, Wang M (2012) Genome shuffling of Streptomyces gilvosporeus for improving natamycin production. J Agric Food Chem 60(23):6026–36
Lv X-A, Jin Y-Y, Li Y-D, Zhang H, Liang X-L (2012) Genome shuffling of Streptomyces viridochromogenes for improved production of avilamycin. Appl Microbiol Biotechnol 97(2):641–8
Mao EF, Lane L, Lee J, Miller JH, Mao EF, Lane L, Lee J, Miller JH (1997) Proliferation of mutators in A cell population. J Bacteriol 179(2):417–22
Miller JH (1996) Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu Rev Microbiol 50:625–43
Muteeb G, Sen R (2010) Random mutagenesis using a mutator strain. Methods Mol Biol 634:411–419
Otte B, Grunwaldt E, Mahmoud O, Jennewein SC-P (2009) Genome shuffling in Clostridium diolis DSM 15410 for improved 1,3-propanediol production. Appl Env Microbiol 75:7610–7616
Patnaik R, Louie S, Gavrilovic V, Perry K, Stemmer WP, Ryan CM, del Cardayré S (2002) GS of Lactobacillus for improved acid tolerance. Nat Biotechnol 20:707–712
Pinel D, D'Aoust F, del Cardayre SB, Bajwa PK, Lee H, Martin VJJC-P (2011) Saccharomyces cerevisiae genome shuffling through recursive population mating leads to improved tolerance to spent sulfite liquor. Appl Env Microbiol 77:4736–4743
Pirakitikulr N, Ostrov N, Peralta-Yahya P, Cornish VW (2010) PCRless library mutagenesis via oligonucleotide recombination in yeast. Protein Sci 19:2336–46
Radding C (1989) Helical RecA nucleoprotein filaments mediate homologous pairing and strand exchange. Biochim Biophys Acta 1008:131–145
Révet B, Sena E, Zarling D (1993) Homologous DNA targeting with RecA protein-coated short DNA probes and electron microscope mapping on linear duplex molecules. J Mol Biol 232:779–791
Sena E, Zarling D (1993) Targeting in linear DNA duplexes with two complementary probe strands for hybrid stability. Nat Genet 3:365–372
Setlow RB (1966) Cyclobutane-type pyrimidine dimers in polynucleotides. Science 153:379–86
Shi D, Wang C, Wang K (2009) Genome shuffling to improve thermotolerance, ethanol tolerance and ethanol productivity of Saccharomyces cerevisiae. J Ind Microbiol Biotechnol 36:139–47
Shi J, Zhang M, Zhang L, Wang P, Jiang L, Deng H (2014) Xylose-fermenting Pichia stipitis by genome shuffling for improved ethanol production. Microb Biotechnol. doi:10.1111/1751-7915.12092
Singer B (1981) Mutagenesis from a chemical perspective: nucleic acid reactions, repair, translation, and transcription. In: Lemontt J, Generoso W (eds) Molecular and cellular mechanisms of mutagenesis. Plenum, New York, pp 1–42
Spencer J, Spencer D (1996) Mutagenesis in yeast. Methods Mol Biol 53:17–38
Tao X, Zheng D, Liu T, Wang P, Zhao W, Zhu M, Jiang X, Zhao Y, Wu X (2012) A novel strategy to construct yeast Saccharomyces cerevisiae strains for very high gravity fermentation. PLoS One 7(2):e31235
Tobin M, Stemmer WPC, Ness JE, Minshull JS (2001) Evolution of whole cells and organisms by recursive sequence recombination. USPTO 6251674B1.
Wang ZG, Wu XH, Friedberg EC (1991) Nucleotide excision repair of DNA by human cell extracts is suppressed in reconstituted nucleosomes. J Biol Chem 266:22472–8
Wang Y, Li Y, Pei X, Yu L, Feng Y (2007) Genome-shuffling improved acid tolerance and l-lactic acid volumetric productivity in Lactobacillus rhamnosus. J Biotechnol 129:510–515
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–8
Wang M, Liu S, Li Y, Xu R, Lu C, Shen Y (2010) Protoplast mutation and genome shuffling induce the endophytic fungus Tubercularia sp. TF5 to produce new compounds. Curr Microbiol 61:254–260
Wang H, Zhang J, Wang X, Qi W, Dai Y (2012a) Genome shuffling improves production of the low-temperature alkalophilic lipase by Acinetobacter johnsonii. Biotechnol Lett 34:145–51. doi:10.1007/s10529-011-0749-7
Wang P-M, Zheng D-Q, Liu T-Z, Tao X-L, Feng M-G, Min H, Jiang X-H, Wu X-C (2012b) The combination of glycerol metabolic engineering and drug resistance marker-aided genome shuffling to improve very-high-gravity fermentation performances of industrial Saccharomyces cerevisiae. Bioresour Technol 108:203–10
Wang C, Wu G, Li Y, Huang Y, Zhang F, Liang X (2013) Genome shuffling of Penicillium citrinum for enhanced production of nuclease P1. Appl Biochem Biotechnol 170:1533–45
Wei P, Li Z, He P, Lin Y, Jiang N (2008) Genome shuffling in the ethanologenic yeast Candida krusei to improve acetic acid tolerance. Biotechnol Appl Biochem 49:113–120
Wei Y, Wang C, Wang M, Cao X, Houa L (2013) Comparative analysis of salt-tolerant gene HOG1 in a Zygosaccharomyces rouxii mutant strain and its parent strain. J Sci Food Agric. doi:10.1002/jsfa.6096
Winston F (2008) EMS and UV mutagenesis in yeast. Curr Protoc Mol Biol Chapter 13:Unit 13.3B. doi: 10.1002/0471142727.mb1303bs82
Xu B, Jin Z, Wang H, Jin Q, Jin X, Cen P (2008) Evolution of Streptomyces pristinaespiralis for resistance and production of pristinamycin by genome shuffling. Appl Microbiol Biotechnol 80:261–267
Xu F, Wang J, Chen S, Qin W, Yu Z, Zhao H, Xing X, Li H (2011) Strain improvement for enhanced production of cellulase in Trichoderma viride. Prikl Biokhim Mikrobiol 47:61–65
Yu L, Pei X, Lei T, Wang Y, Feng Y (2008) Genome shuffling enhanced l-lactic acid production by improving glucose tolerance of Lactobacillus rhamnosus. J Biotechnol 134:154–159
Yu G, Hu Y, Chen L, Wang L, Liu N, Yin Y, Zhao J (2014) Genome shuffling of Streptomyces roseosporus for improving daptomycin production. Appl Biochem Biotechnol. doi:10.1007/s12010-013-0687-z
Zhang W, Geng A (2012) Improved ethanol production by a xylose-fermenting recombinant yeast strain constructed through a modified genome shuffling method. Biotechnol Biofuels 5(1):46
Zhang Y-XX, Perry K, Vinci VA, Powell K, Stemmer WPC, del Cardayré SB (2002) Genome shuffling leads to rapid phenotypic improvement in bacteria. Nature 415:644–6
Zhang Y, Liu J-Z, Huang J-S, Mao Z-W (2010) Genome shuffling of Propionibacterium shermanii for improving vitamin B12 production and comparative proteome analysis. J Biotechnol 148:139–43
Zhang J, Wang X, Diao J, He H, Zhang Y, Xiang W (2013) Streptomycin resistance-aided genome shuffling to improve doramectin productivity of Streptomyces avermitilis NEAU1069. J Ind Microbiol Biotechnol 40:877–89
Zhao K, Ping W, Zhang L, Liu J, Lin Y, Jin T, Zhou D (2008) Screening and breeding of high taxol producing fungi by genome shuffling. Sci China C Life Sci 51:222–231
Zhao J, Li Y, Zhang C, Yao Z, Zhang L, Bie X, Lu F, Lu Z (2012) Genome shuffling of Bacillus amyloliquefaciens for improving antimicrobial lipopeptide production and an analysis of relative gene expression using FQ RT-PCR. J Ind Microbiol Biotechnol 39:889–96
Zheng H, Gong J, Chen T, Chen X, Zhao X (2010) Strain improvement of Sporolactobacillus inulinus ATCC 15538 for acid tolerance and production of d-lactic acid by genome shuffling. Appl Microbiol Biotechnol 85:1541–1549
Zheng D-Q, Wu X-C, Tao X-L, Wang P-M, Li P, Chi X-Q, Li Y-D, Yan Q-F, Zhao Y-H (2011a) Screening and construction of Saccharomyces cerevisiae strains with improved multi-tolerance and bioethanol fermentation performance. Bioresour Technol 102:3020–7
Zheng DQ, Wu XC, Wang PM, Chi XQ, Tao XL, Li P, Jiang XH, Zhao YH (2011b) Drug resistance marker-aided genome shuffling to improve acetic acid tolerance in Saccharomyces cerevisiae. J Ind Microbiol Biotechnol 38:415–422
Zheng P, Liu M, Liu X, Du Q-Y, Ni Y, Sun Z-H (2012) Genome shuffling improves thermotolerance and glutamic acid production of Corynebacteria glutamicum. World J Microbiol Biotechnol 28:1035–43
Zheng D-Q, Chen J, Zhang K, Gao K-H, Li O, Wang P-M, Zhang X-Y, Du F-G, Sun P-Y, Qu A-M, Wu S, Wu X-C (2013a) Genomic structural variations contribute to trait improvement during whole-genome shuffling of yeast. Appl Microbiol Biotechnol. doi:10.1007/s00253-013-5423-7
Zheng P, Zhang K, Yan Q, Xu Y, Sun Z (2013b) Enhanced succinic acid production by Actinobacillus succinogenes after genome shuffling. J Ind Microbiol Biotechnol 40:831–40
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Biot-Pelletier, D., Martin, V.J.J. Evolutionary engineering by genome shuffling. Appl Microbiol Biotechnol 98, 3877–3887 (2014). https://doi.org/10.1007/s00253-014-5616-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5616-8