
Abstract
The double-stranded RNA (dsRNA) mediated RNA interference (RNAi) is widely employed in silkworm and its tissue-derived cell lines for gene function analysis. Baculovirus expression vector system (BEVS) has an advantage for large-scale protein expression. Previously, combining these useful tools, we improved traditional AcMNPV-Sf9 BEVS to produce modified target glycoproteins, where the ectopic expression of Caenorhabditis elegans systemic RNAi defective-1 (SID-1) was found to be valuable for soaking RNAi. In current study, we applied CeSID-1 protein to a Bombyx mori NPV (BmNPV)-hypersensitive Bme21 cell line and investigated its properties both in soaking RNAi ability and recombinant protein expression. The soaking RNAi-mediated suppression in the Bme21 cell enables us to produce modified glycoproteins of interest in BmNPV–Bme21 BEVS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Baculovirus/insect cell expression vector systems (BEVSs) are widely employed for large-scale functional glycoprotein productions due to the relatively similar post-translational protein modifications (PTMs) in most insect cells (O'Reilly et al. 1992; Hitchman et al. 2009). The Autographa californica multiple nucleopolyhedrovirus (AcMNPV) is the most developed pathogen used for infecting its hypersensitive cells such as Sf9, Sf21, and Hi-5 cell lines (McCall et al. 2005). While another baculovirus, the Bombyx mori nucleopolyhedrovirus (BmNPV) is not frequently utilized partly because few host cell lines are available for producing proteins with high yield. The B. mori tissue-derived cell lines, such as BmN, BmN4, and Bm5, were moderately productive in mass production of foreign proteins (Lee et al. 2012), although silkworm cell lines maintained in different laboratories often show different BmNPV sensitivities (Katsuma and Shimada 2012). Previously, we identified a BmNPV high permissive cell lines Bme21 and observed a ~100-fold more reporter protein production than that from BmN4 and Bm5 cells when infected by BmNPV (Lee et al. 2012).
In the past decade, sequence-specific RNA interference (RNAi) using double-stranded RNA (dsRNA) or small interference RNA (siRNA) has been widely employed as a effective tool for suppressing specific gene expression (Mohr et al. 2010). In Caenorhabditis elegans, multispan transmembrane protein systemic RNAi defective-1 (SID-1) was reported to be essential for the uptake of long dsRNA into cell across the cytoplasmic membrane (Feinberg and Hunter 2003; Shih et al. 2009; McEwan et al. 2012). Unlike C. elegans and Drosophila, most of Lepidopteran insects and cells have no capability of autonomous dsRNA uptake (Feinberg and Hunter 2003; Terenius et al. 2011). Thus, RNAi is typically carried out by the transfection of long dsRNAs into these insect cells (Terenius et al. 2011). To overcome the shortcomings caused by transfection reagents, CeSID-1 has been introduced to several model organisms to stimulate the spontaneous uptake of long dsRNAs (Feinberg and Hunter 2003; Tsang et al. 2007; Mon et al. 2012; Kobayashi et al. 2012; Xu et al. 2013). These established cell lines were sensitive to soaking RNAi. Previously, we have established a BmN4–SID1 cell line for soaking RNAi (Mon et al. 2012). This B. mori-derived cell line is particularly useful for lepidopteran gene analysis since its genome sequence is available (The International Silkworm Genome Consortium 2008). The BmNPV sensitivity and protein expression level of the BmN4–SID1 cell by BEVS, however, was moderate as expected from the nature of its origin.
In the current study, we generated a novel Bme21–SID1 cell line in which the soaking RNAi selectively depleted the expression of endogenous and exogenous genes of interest (Lee et al. 2012). The Bme21 cell line is distinct from other B. mori tissue-derived cell lines not only in cell morphology but also in its response to baculovirus infection (Lee et al. 2012). These novel characteristics make this line an attracted platform for modification and mass production of foreign proteins by BEVS.
Materials and methods
Plasmids constructions and generation of stable cell line
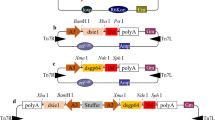
The full-length cDNA sequence of CeSID-1 was a kind gift from Dr. Kobayashi (National Institute of Agrobiological Sciences, Japan). The piggyBac-based transposition vector, pBac-IE2-CeSID1_DsRedpuro was constructed by Gateway reaction as described previously (Xu et al. 2013). The CeSID-1 and DsRED-Puro genes are inserted in the same piggyBac cassette and expressed under the control of Orgyia pseudotsugata IE2 (OpIE2) and OpIE1 promoters, respectively (Fig. 1a). Then it was co-transfected with a piggyBac helper plasmid into Bme21 cells, followed by selection with the antibiotic Puromycin to establish stable cell lines. The Puromycin selected cells were further cloned in 96-well plates (Falcon, BD Biosciences).

a Schematic representation of piggyBac-based transposition vector, pBac-IE2-CeSID1_DsRedpuro. b Schematic representation of recombinant BmNPV/T3. The plasmid pDEST8-polh-Target-tev-H8 was generated from its corresponding pENTR11-L21-Target-tev-H8 and pDEST8 (Invitrogen) vectors by Gateway LR clonase reaction (Invitrogen). Subsequently, bacmid DNAs and BmNPV P3 viruses were obtained using the methods as described in the “Materials and methods” section
Insect cell culture and baculoviruses
The B. mori BmN4, BmN4–SID1, Bme21, and Bme21–SID1 cells were maintained at 27 °C on IPL-41 (Sigma) supplemented with 10 % fetal bovine serum (FBS, Gibco). B. mori cell lines BmN4 and Bme21 were provided from Dr. Chisa Aoki of Kyushu University Graduate School. The Bme21 cell is available upon the request to the Kyushu University Material Management Center (http://mmc-u.jp/en/doc/howto/). To produce target glycoproteins, dsRNA-soaked Bme21–SID1 cells were adapted into a serum-free medium (Cosmedium 009, CosmoBio) before viral infection.
The recombinant BmNPVs, BmNPV-polh-Luciferase-tev-H8, BmNPV-polh-30K6G–mDsRED-tev-H8, BmNPV-polh-30K6G–PSTBP3-tev-H8 (PSTBP3: Takifugu rubripes Pufferfish saxitoxin- and tetrodotoxin-binding protein 3) and BmNPV-polh-30K6G–EPO-tev-H8 (human erythropoietin) were constructed in our previous studies (Xu et al. 2013; Nagata et al. 2013). The silkworm-derived 30K6G N-terminal sequence is employed as a signal peptide in secreted mDsRed, PSTBP3, and EPO proteins. For the recombinant BmNPV strains expressing nonsecreted mCherry protein, pDEST8-polh-mCherry-tev-H8 was generated from its corresponding pENTR11-L21-TEV-H8 (L21 a lobster L21 sequence for enhancing translation efficiency, TEV tobacco etch virus protease cleavage site, and H8 a poly-histidine tag at the C-terminus; Soejima et al. 2013) and pDEST8 (Invitrogen) vectors by Gateway LR clonase reaction (Invitrogen). Subsequently, bacmid DNAs and BmNPV P3 viruses were obtained using the methods as described previously (Motohashi et al. 2005; Lee et al. 2012). Briefly, Escherichia coli BmT3DH10Bac was transformed with the pDEST8-Target-tevH8 plasmid. Positive colonies containing the bacmid for expression of target protein were further cultured and the bacmid DNA was isolated using the QIAGEN large-construct kit. The bacmid was subsequently transfected into Bme21 cells using Fugene HD (Promega) to yield recombinant BmNPV/T3 virus. Detailed procedures for plasmid constructions are illustrated in Fig. 1b. The viral titers were determined by the end-point dilution method (O'Reilly et al. 1992).
dsRNA synthesis and RNAi
For dsRNA synthesis, all the sequences were amplified from the first-strand cDNA or BmT3DH10Bac bacmid DNA using the gene-specific primers listed in Table 1, and cloned into a pLits vector containing a dual T7 promoter (Li et al. 2012). The dsRNAs for EGFP, Luciferase, BmNPV-IE1, BmRNRS, BmCDK1, BmKIF23, BmFDL, BmIAP1, and BmIAP2 were synthesized by in vitro transcription as described previously (Li et al. 2012). The soaking RNAi of Bme21 and Bme21–SID1 cells were performed as described previously (Mon et al. 2012).
RNA extraction and semiquantitative PCR
Total RNA from Bme21 cells was isolated and reversed according to the procedures described previously (Xu et al. 2013). Semiquantitative RT-PCR was performed routinely as described except for the gene-specific primers used that are listed in Table 1. The B. mori glyceraldehyde-3-phosphate dehydrogenase (BmGAPDH) mRNA was used as internal control. The expression levels of the target genes were quantitated by ImageJ software and normalized to the BmGAPDH levels.
Viral infection and luciferase activity assay
BmN4 or Bme21 cells (5.0 × 104 per well) in 24-well plates were plated and infected with BmNPV-polh-Luc or BmNPV-polh-mCherry at multiplicities of infection (MOI) of 0.1. As for soaking RNAi in Bme21–SID1 cells, dsRNAs were added immediately after cell plating at a concentration of 100 ng/ml. Two days after dsRNA soaking (for BmIAP1, RNAi soaking for 1 day), the cells were infected with BmNPV-polh-Luc at MOI of 0.1 with additional 100 ng/ml dsRNA. The infected cells were harvested at 72 h post-infection (hpi), and subjected to luciferase assay as described previously (Lee et al. 2012). BmNPV-polh-mCherry-infected cells were then observed under a Biozero fluorescence microscope (Keyence) for confirming the mCherry protein expressions and the cell medium were further used for titer determinations.
Fluorescence microscopy
The Bme21 or Bme21–SID1 cells soaked by dsRNA (100 ng/ml) against EGFP, RNRS, CDK1, or KIF23 for 4 days were harvested and washed twice with ice-cold PBS. The slides were subsequently prepared by cytospinning the samples (cytospin4; Thermo Scientific) at 1,000 rpm for 5 min and further fixed in methanol at −20 °C for 30 min. The fixed cells were then stained with DAPI (0.1 μg/ml; Invitrogen) for 30 min and mounted with VECTASHIELD hardset mounting medium (Vector Labs). Fluorescent images were obtained using a Biozero fluorescence microscope (Keyence) for observation of the arrested cell cycle progression by loss of RNRS, CDK1, and KIF23, respectively.
Western blotting
Cell lysate (nonsecreted protein) or culture medium (secreted protein) from baculovirus-infected cells was harvested at 72 hpi. Samples treated with peptide-N-glycosidase F (PNGase F, New England Biolabs) were handled according to the manufacturer's guidelines. After centrifugation, 10 μl protein samples were subjected to SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore), and further blocked in 0.5 % skim milk (Wako). Subsequently, the His-tagged proteins were detected with HisProbe-HRP (1:30,000 in 0.5 % skim milk; Pierce) followed by chemiluminescence using SuperSignal West Pico substrate (Pierce) as instructed by the manufacturer. The band signals were quantified and normalized to their control signals with ImageJ software.
Nucleotide sequence accession number
Nucleotide sequence data of several genes used in this study are provided with the following accession numbers: NC_001962.1 (BmNPV-IE1), AB703263 (BmRNRS), NM_001044047 (BmCDK1), XM_004930714.1 (BmKIF23), AB540233 (BmFDL), NM_001043559 (BmIAP1), and NM_001202529 (BmIAP2).
Results
Bme21 cell line is hypersensitive to BmNPV
We have reported previously that Bme21 cell line is hypersensitive to BmNPV infection (Lee et al. 2012). It is known that cultured cell line undergoes subtle changes during continuous maintaining and would display divergent properties such as virus susceptibility (Lorenzen et al. 1999; Hiorns et al. 2004). Thus, in the current study, we first confirmed the susceptibility of Bme21 cells to BmNPV infections, and BmN4 is employed as a moderately sensitive control. We utilized two reporter genes, luciferase and mCherry, to analyze the very late gene expression of viral infections, which were demonstrated in Fig. 2 (a: luciferase, b: mCherry). Compared with infected BmN4 cells, Bme21 showed prominently high reporter gene expression levels. Subsequently, we determined the titers of budded viruses (BVs) produced from the BmNPV-polh-mCherry-infected BmN4 and Bme21 cells (Fig. 2c). Consistent with the results from the reporter assay, significantly higher BVs were produced from Bme21 cells, indicating the stable hypersensitivity of Bme21 to the BmNPV infection.

Bme21 cell line is hypersensitive to BmNPV. BmN4 or Bme21 cells (5.0 × 104) in 24-well plates were infected with BmNPV-polh-Luc or BmNPV-polh-mCherry at multiplicities of infection (MOI) of 0.1, respectively. The relative luciferase activity (a) or mCherry fluorescence (b) at 48 hpi is plotted for both cell lines. The luciferase activity was normalized to the mean of luciferase activity from BmN4 cell line. Compared with infected BmN4 cells, Bme21 showed prominently high reporter gene expression levels, indicating the stable hypersensitivity of Bme21 to the BmNPV infection. c Titers of budded viruses (BVs) produced from the BmNPV-polh-mCherry-infected BmN4 and Bme21 cells were determined in Bme21 cells by the end-point dilution method
Generation of a stable Bme21 cell line overexpressing CeSID-1
As a transmembrane protein, CeSID-1 contains a large N-terminal extracellular domain (ECD) followed by 11 transmembrane regions (Feinberg and Hunter 2003). We reported previously that N-terminal fused tag could affect CeSID-1 function for dsRNA uptake (Mon et al. 2012). In this study, we also chose to overexpress CeSID-1 without peptide tag. Thus, we generated a transgenic Bme21-CeSID1–DsRed-Puro (hereafter referred to as Bme21–SID1) cell line as described under the “Materials and methods” section. To ensure the stable CeSID-1 expression, we further performed cell cloning in 96-well plate by continuous antibiotic selection (Puromycin) and red fluorescence validation under the microscope (data not shown). The generated cell lines were normally passaged at higher split ratios until it is in decent cell condition and subsequently employed for soaking RNAi experiments.
Ectopic CeSID-1 expression allows efficient soaking RNAi in Bme21 cells
To investigate whether CeSID-1 could function as dsRNA receptor in Bme21 cells, we investigated the soaking RNAi efficacy of Bme21–SID1 cells by selectively suppressing three distinct cell cycle-related factors, Ribonucleoside diphosphate reductase small subunit (RnrS), cyclin-dependent kinase 1 (CDK1) and kinesin-like protein KIF23. It has been reported that depletion of RNRS leads to cell cycle arrest at S phase and induces apoptosis (Björklund et al. 2006), while RNAi for CDK1 causes mitotic catastrophe and induction of cell death (Björklund et al. 2006) and suppression of KIF23 results in a high frequency of binucleate cells (Goshima et al. 2007). As shown in Fig. 3a, soaking RNAi of these cell cycle factors in Bme21–SID1 cells triggered severe loss-of-function phenotypes of large cell population at 4 days of soaking (100 ng/ml dsRNA). From Fig. 3b, through RT-PCR, we confirmed that the mRNA expression levels of these three genes were knocked down to approximately 20 % only when corresponding dsRNA was added, indicating the efficient gene-specific RNAi was activated after direct dsRNA soaking in Bme21–SID1 cells.

Bme21–SID1 cell lines can be utilized to repress either endogenous or exogenous gene expression. a Three cell cycle-related genes, BmRNRS, BmCDK1, and BmKIF23, were used to investigate the efficacy of soaking RNAi against endogenous target genes. RNAi phenotypes and mRNA expressions were investigated at 4 days after RNAi induction to Bme21–SID1 cells. To visualize the mitotic catastrophe caused by the gene depletion, the nuclei were stained by DAPI dye. Arrows indicate the gene specifically induced arrest of cell cycle progression. b Efficient gene-specific RNAi was confirmed by semiquantitative RT-PCR using the same samples with (a). The dsEGFP treatment was used as a negative control and BmGAPDH mRNA were used as expression standard. All these images were finally adjusted to maximize visualization by ImageJ. c The plasmid pIE2-Luciferase was employed for transient expressions of luciferase in Bme21–SID1 cells. After transfection, the cells were soaked by dsRNA against EGFP or luciferase for 48 h. Cells were subsequently lysed and subjected to luciferase assay to determine the relative expressions. The normal Bme21 cells (negative control) were treated under the same condition and the results were illustrated in d, e and f, respectively. All values represent the mean ± SD for N = 6 independent infections. Indicated P value was assessed by Student's t test (n.s. not significant, P > 0.05; ***P < 0.001). See “Materials and methods” section for more details
To validate its competency for soaking RNAi targeting exogenous genes, we expressed luciferase reporter gene in Bme21–SID1 cells by the transient transfection of pIE2-Luc. Then cells were soaked simultaneously with dsRNA against EGFP (negative control) or luciferase. As shown in Fig. 3c, the significant knockdown of luciferase gene expression were observed in the presence of dsLuc, suggesting that our system is well suited for the control of exogenous genes. Meanwhile, the normal Bme21 cells were treated under the same conditions and investigated as negative controls (Fig. 3d, e, f). As expected, almost no dsRNA uptake property was detected in these dsRNA-soaking analyses.
It is noteworthy, however, that many cloned cell lines were low sensitive to soaking RNAi and some colonies even displayed no affect when 1 μg/ml dsRNA was used (data not shown). Semiquantitative RT-PCR analysis confirmed that there were no substantial differences in the mRNA expression levels of CeSID-1 among the dsRNA-sensitive and dsRNA -insensitive Bme21–SID1 cells (Fig. 4).

Identification of mRNA expression levels between dsRNA-sensitive and dsRNA-insensitive Beme21–SID1 colonies. Each three dsRNA-sensitive and dsRNA-insensitive cells were employed to analyze the mRNA expression levels of CeSID-1 and Bme21 cells was used as negative control. The procedures for RNA isolation and cDNA synthesis were described in the “Materials and methods” section. Semiquantitative RT-PCR was performed routinely as described except for the CeSID-1 gene-specific primers used that are listed in Table 1. The Bombyx mori glyceraldehyde-3-phosphate dehydrogenase (BmGAPDH) mRNA was used as internal control
Comparison analysis of soaking RNAi against BmIAP1 between Bme21–SID1 and BmN4–SID1 cell lines
It has reported that knockdown of B. mori cellular inhibitor of apoptosis 1 (BmIAP1) induces death programs for apoptosis in BmN cells (Suganuma et al. 2011). To investigate BmIAP1 function in distinct cell lines, we employed our two soaking RNAi-sensitive cell lines, BmN4–SID1 and Bme21–SID1. At 24 h of soaking RNAi against BmIAP1, Bme21–SID1 cells demonstrated rapid induction of apoptosis (Fig. 5a), while almost no apoptotic phenotype was observed in BmN4–SID1 cell lines with the same time point. When subsequently infected with BmNPV-polh-Luc, dsBmIAP1-soaked Bme21 cells produced less luciferase reporter mainly due to that most of the Bme21–SID1 cells had already undergone the process of apoptosis (Fig. 5b). At 72 h of soaking RNAi, severe apoptosis phenotype can be also observed in BmN4 cells, which further reveals the conserved function of BmIAP1 as an inhibitor of apoptosis in B. mori cell lines.

Bme21–SID1 cells demonstrate rapid response upon the depletion of BmIAP1. BmN4–SID1 and Bme21–SID1 cell lines were used for soaking RNAi against BmIAP1. a At 24 h of soaking, two cell lines (left panel: BmN4–SID1; right panel: Bme21–SID1) were observed under microscope. Arrows indicate the apoptotic phenotype induced by the depletion of BmIAP1. b The dsBmIAP1-soaked cells were subsequently infected with BmNPV-polh-Luc. Cell lysates were subjected to luciferase assay to determine the relative expressions, which were normalized to that in dsEGFP-treated cells, respectively. 68.4 % and 31.8 % indicate the percentage of downgrade in each cell line. All values represent the mean ± SD for N = 6 independent infections
BmNPV/Bme21–SID1 BEVS is a useful platform for protein production
Prior to the application of BmNPV/Bme21–SID1 system, we evaluated whether ectopic expression of CeSID-1 could affect the viral infection and replication. As shown in Fig. 6a, no significant differences were observed in the production of luciferase reporter gene between CeSID-1 negative and positive cells, indicating no involvement of CeSID-1 in the interaction between Bme21 and BmNPV. Subsequently, we test our cell lines with dsRNA against one of the essential genes for BmNPV replication, immediate-early IE1 (Bm-IE1). When simultaneously soaked with dsEGFP (negative control), dsLuc (positive control), and dsIE1, as expected, only dsLuc- and dsIE1-treated cells remarkably reduced the viral replications as determined by the luciferase gene expressions (Fig. 6b).

Bme21–SID1 cell line can be utilized to identify BmNPV gene functions. a Ectopic expression of CeSID-1 does not affect the Bme21 cell susceptivity to BmNPV infection. The same numbers of Bme21 or Bme21–SID1 cells were infected with BmNPV-polh-Luc at the same MOI of 0.1. The fold change of relative luciferase activity at 48 hpi is plotted for both cell lines. b The Bme21–SID1 cells were preincubated with dsRNA against EGFP, Luciferase or IE1 and subsequently infected with BmNPV-polh-Luc at MOI of 0.1. The fold changes of relative luciferase activity were analyzed. Soaking RNAi of viral IE1 and Luciferase affects luciferase gene expressions. All values represent the mean ± SD for N = 6 independent infections. Indicated P value was assessed by Student's t test (n.s. not significant, P > 0.05; ***P < 0.001)
Since we have reported in BmN4 cells that the depletion of B. mori β-N-acetylglucosaminidase (BmFDL) converted paucimannosidic structures into complex-type structures of glycoproteins (Nagata et al. 2013), we then investigated whether modified N-glycosylated glycoprotein could be produced through BmNPV–Bme21 BEVS. In the current study, we used mDsRed with signal peptide (no N-glycosylation site), and PST-BP3 (6 N-glycosylation sites) and human EPO (3 N-glycosylation sites) as reporters to analyze the effect of soaking RNAi against BmFDL. By RT-PCR, we confirmed that about 84 % mRNA expression of BmFDL was suppressed after soaking RNAi against BmFDL in Bme21–SID1 cells (Fig. 7a). We compared secreted PST-BP3 products from BmN4–SID1 and Bme21–SID1 cells with or without the depletion of BmFDL (Fig. 7b). Since FDL functions conservatively in lepidopteran cells (Geisler et al. 2008; Nagata et al. 2013; Xu et al. 2013), we validated that these two silkworm cells generate the same types of N-lined glycoproteins and Bme21–SID1 cells yield much more than that from BmN4–SID1 cells. We then analyzed the N-glycan structures attached to the secreted EPO at 72 hpi with or without peptide-N-glycosidase F digestion to remove N-linked glycans. As shown in Fig. 7c, consistent with our previous studies (Nagata et al. 2013; Xu et al. 2013), the Bme21–SID1 cells produced EPO protein with higher molecular weight of N-glycans by FDL depletion, indicating the complex-type N-linked glycan could be also restored after FDL depletion in Bme21 cells.

BmNPV–Bme21–SID1 cell expression system can be utilized to generate modified N-glycosylated glycoproteins. a Soaking RNAi-mediated BmFDL suppression was validated by semiquantitative RT-PCR. The expression levels of the BmFDL were quantitated by ImageJ software and normalized to the BmGAPDH levels. b Loss of BmFDL in BmN4 and Bme21 cells leads to same N-glycosylated PSTBP3 proteins. The BmN4–SID1 and Bme21–SID1 cells were preincubated with dsEGFP or dsFDL and subsequently infected with recombinant BmNPV–PSTBP3. All products were analyzed by western blot. c Knockdown of endogenous BmFDL leads to a band shift of the EPO protein. The Bme21–SID1 cells were preincubated with dsEGFP or dsFDL, and subsequently infected with recombinant BmNPV–mDsRed or BmNPV –EPO. Moreover, all secreted products were digested with peptide-N-glycosidase F to remove N-linked glycans. Accordingly, an apparent increase of the electrophoretic mobility was detected in EPO (the right panel) but not in mDsRed (the left panel) protein samples
Discussion
Together with our current report, six distinct cell lines have provided evidence for CeSID-1-dependent passive dsRNA uptakes (Feinberg and Hunter 2003; Tsang et al. 2007; Mon et al. 2012; Kobayashi et al. 2012; Xu et al. 2013). We tested our cloned transgenic cell lines by soaking with dsRNA against either endogenous or exogenous gene targets. The present results showed that ectopic expression of CeSID-1 enabled efficient soaking RNAi in Bme21 cells. It is interesting to find that some cloned lines demonstrated low sensitivity to soaking RNAi, possibly due to the low expression amount of CeSID-1. It has been reported that no significant enhancements of dsRNA uptake were detected in CeSID-1 transgenic silkworms (Kobayashi et al. 2012). This may suggest that a high expression level of SID-1 is crucial for the support of systemic RNAi. Furthermore, other SID protein family members, such as SID-2 and SID-5, have also been identified as SID-1 associated proteins required for efficient systemic RNAi (McEwan et al. 2012; Hinas et al. 2012). Thus, our observations of different soaking RNAi efficacies in CeSID-1 transgenic Bme21 cell clones may indicate that the endogenous silkworm SID protein family members are also important for the function of exogenous CeSID-1 as a sufficient dsRNA channel.
Herein we observed the relatively rapid induction of apoptosis in BmNPV-hypersensitive Bme21 cells treated with dsRNA against BmIAP1. No significant difference on the mRNA expression of BmIAP1 between BmN4 and Bme21 cells (online resource 1a) implies the differential sensitivities of these two cells upon the depletion of BmIAP1. The silkworm possesses two functional IAP gene homologs, BmIAP1 and BmIAP2, which consist of one RING domain and two or three BIR domains, respectively (Zhang et al. 2010). It is reported in Drosophila that loss of DIAP1 but not IAP2 leads to apoptosis and IAP2 is dispensable for cell viability in haemocyte-like cells (Gesellchen et al. 2005). We also confirmed in Bme21 cells that loss of BmIAP2 neither induces apoptosis nor affects baculoviral replications according to the luciferase gene expressions (online resource 1b). The IAP protein family is identified as one of the homologs shared by both baculovirus and host (Katsuma et al. 2008). Since the apoptosis is vital in the host immune response to baculovirus infections (Ikeda et al. 2013), our results provide insight into the anti- or pro-viral analysis of IAP protein family in these two different cell lines.
Previously, using Sf9–SID1 cell lines we tested the soaking RNAi method for the AcMNPV gene function analysis (Xu et al. 2013). In this paper, we identified the function of BmNPV IE1 gene, which is essential for viral replication. The BmNPV and AcMNPV are closely related sharing about 93 % identities on amino acid sequences (Gomi et al. 1999). Despite their similarity, they differ significantly in their infectivity spectrum (Katou et al. 2006). Not only viral genes but also host factors are reported to be involved in their replications (Ikeda et al. 2013). Nonetheless, it is still elusive to explain the underlying mechanisms of host response to baculovirus infections. Our CeSID-1 transgenic cell lines including Sf9–SID1, BmN4–SID1, and Bme21–SID1 lines would provide a fine platform for high-throughput RNAi-based investigation on comparative virus–host interactions.
Owing to that, Lepidopteran cells are mainly of greatest concern in the protein productions, it is vital to establish a stable platform in either host- or baculovirus-dependent manner to provide both high yields and high qualities of produced proteins. Similar to silkworm strains, the tissue-derived silkworm cell lines show particular cytological characteristics and also demonstrate diverse responses to microbial infections such NPVs (Bao et al. 2009; Lee et al. 2012; Xue at al. 2012). On the basis of our previous study, Bme21 is the most productive silkworm cells to the BmNPV infection, while BmN4 or Bm5 cells were found moderately sensitive (Lee et al. 2012) This is one of the hurdles of BmN4/Bm5 cells to be used for expressing foreign proteins in a high yield way. To our knowledge, Bme21 cells are by far the finest cell host for the establishment of BmNPV-dependent BEVS.
In contrast to mammalian N-glycosylation pathway, the terminal N-acetylglucosamine residue of GlcNAcMan3GlcNAc2-R is trimmed by β-N-acetylglucosaminidase (FDL) to generate Man3GlcNAc2-R (Altmann et al. 1995; Léonard et al. 2006; Geisler et al. 2008; Nagata et al. 2013). Thus, glycoproteins produced using insect baculovirus lack complex-type N-glycans with complete sialylated antennate, which therefore makes insects unsuitable for future pharmaceutical applications (Watanabe et al. 2002). To data, scarce reports have been found describing N-glycosylation pathways of silkworm and its cell lines. Herein we confirmed in Bme21 cells that complex-type N-linked glycan could be also restored after the FDL depletion. Since Bme21–SID1 cell is highly sensitive to both BmNPV infection and soaking RNAi, we could produce different types of glycoproteins in high yields by soaking RNAi against specific PTM enzymes. On the other hand, taken together with our results in BmN4 cells, we considered that solely RNAi of BmFDL is inadequate for generating complete mammalian-like N-linked glycans in the baculovirus insect cell system (Nagata et al. 2013). During the last three decades, considerable efforts have been made for humanizing insect cells to produce complex type N-glycans. Generally, it was clear that β-1,2-N-acetylglucosaminyltransferase I (GnTI), β-1,2-N-acetylglucosaminyltransferase II (GnTII), β-1,4-galactosyltransferase, and sialyltransferases were the crucial enzymes limiting the glycan structures in insect cells (Jarvis et al. 1998). Combination of these exogenous genes expression and soaking RNAi against the genes that act negatively in glycosylation or secretion pathways may be useful for mass production of glycoproteins.
References
Altmann F, Schwihla H, Staudacher E, Glössl J, März L (1995) Insect cells contain an unusual, membrane-bound beta-N-acetylglucosaminidase probably involved in the processing of protein N-glycans. J Biol Chem 270:17344–17349
Bao YY, Tang XD, Lv ZY, Wang XY, Tian CH, Xu YP, Zhang CX (2009) Gene expression profiling of resistant and susceptible Bombyx mori strains reveals nucleopolyhedrovirus-associated variations in host gene transcript levels. Genomics 94:138–145
Björklund M, Taipale M, Varjosalo M, Saharinen J, Lahdenperä J, Taipale J (2006) Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature 439:1009–1013
Feinberg EH, Hunter CP (2003) Transport of dsRNA into cells by the transmembrane protein SID-1. Science 301:1545–1547
Geisler C, Aumiller JJ, Jarvis DL (2008) A fused lobes gene encodes the processing beta-N-acetylglucosaminidase in Sf9 cells. J Biol Chem 283:11330–11339
Gesellchen V, Kuttenkeuler D, Steckel M, Pelte N, Boutros M (2005) An RNA interference screen identifies inhibitor of Apoptosis Protein 2 as a regulator of innate immune signalling in Drosophila. EMBO Rep 6:979–984
Gomi S, Majima K, Maeda S (1999) Sequence analysis of the genome of Bombyx mori nucleopolyhedrovirus. J Gen Virol 80(Pt 5):1323–1337
Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, Stuurman N (2007) Genes required for mitotic spindle assembly in Drosophila S2 cells. Science 316:417–421
Hinas A, Wright AJ, Hunter CP (2012) SID-5 is an endosome-associated protein required for efficient systemic RNAi in C. elegans. Curr Biol 22:1938–1943
Hiorns LR, Bradshaw TD, Skelton LA, Yu Q, Kelland LR, Leyland-Jones B (2004) Variation in RNA expression and genomic DNA content acquired during cell culture. Br J Cancer 90:476–482
Hitchman RB, Possee RD, King LA (2009) Baculovirus expression systems for recombinant protein production in insect cells. Recent Pat Biotechnol 3:46–54
Ikeda M, Yamada H, Hamajima R, Kobayashi M (2013) Baculovirus genes modulating intracellular innate antiviral immunity of lepidopteran insect cells. Virology 435:1–13
Jarvis DL, Kawar ZS, Hollister JR (1998) Engineering N-glycosylation pathways in the baculovirus-insect cell system. Curr Opin Biotechnol 9:528–533
Katou Y, Ikeda M, Kobayashi M (2006) Abortive replication of Bombyx mori nucleopolyhedrovirus in Sf9 and high five cells: defective nuclear transport of the virions. Virology 347:455–465
Katsuma S, Shimada T (2012) Comparative studies of Bombyx mori nucleopolyhedrovirus infection in BmN4 cell lines maintained in different laboratories. J Insect Biotechnol Sericology 81:7–12
Katsuma S, Kawaoka S, Mita K, Shimada T (2008) Genome-wide survey for baculoviral host homologs using the Bombyx genome sequence. Insect Biochem Mol Biol 38:1080–1086
Kobayashi I, Tsukioka H, Kômoto N, Uchino K, Sezutsu H, Tamura T, Kusakabe T, Tomita S (2012) SID-1 protein of Caenorhabditis elegans mediates uptake of dsRNA into Bombyx cells. Insect Biochem Mol Biol 42:148–154
Lee JM, Kawakami N, Mon H, Mitsunobu H, Iiyama K, Ninaki S, Maenaka K, Park EY, Kusakabe T (2012) Establishment of a Bombyx mori nucleopolyhedrovirus (BmNPV) hyper-sensitive cell line from the silkworm e21 strain. Biotechnol Lett 34:1773–1779
Léonard R, Rendic D, Rabouille C, Wilson IBH, Préat T, Altmann F (2006) The Drosophila fused lobes gene encodes an N-acetylglucosaminidase involved in N-glycan processing. J Biol Chem 281:4867–4875
Li Z, Tatsuke T, Sakashita K, Zhu L, Xu J, Mon H, Lee JM, Kusakabe T (2012) Identification and characterization of polycomb group genes in the silkworm, Bombyx mori. Mol Biol Rep 39:5575–5588
Lorenzen E, Carstensen B, Olesen NJ (1999) Inter-laboratory comparison of cell lines for susceptibility to three viruses: VHSV, IHNV and IPNV. Dis Aquat Org 37:81–88
McCall EJ, Danielsson A, Hardern IM, Dartsch C, Hicks R, Wahlberg JM, Abbott WM (2005) Improvements to the throughput of recombinant protein expression in the baculovirus/insect cell system. Protein Expr Purif 42:29–36
McEwan DL, Weisman AS, Hunter CP (2012) Uptake of extracellular double-stranded RNA by SID-2. Mol Cell 47:746–754
Mohr S, Bakal C, Perrimon N (2010) Genomic screening with RNAi: results and challenges. Annu Rev Biochem 79:37–64
Mon H, Kobayashi I, Ohkubo S, Tomita S, Lee J, Sezutsu H, Tamura T, Kusakabe T (2012) Effective RNA interference in cultured silkworm cells mediated by overexpression of Caenorhabditis elegans SID-1. RNA Biol 9:40–46
Motohashi T, Shimojima T, Fukagawa T, Maenaka K, Park EY (2005) Efficient large-scale protein production of larvae and pupae of silkworm by Bombyx mori nuclear polyhedrosis virus bacmid system. Biochem Biophys Res Commun 326:564–569
Nagata Y, Lee JM, Mon H, Imanishi S, Hong SM, Komatsu S, Oshima Y, Kusakabe T (2013) RNAi suppression of β-N-acetylglucosaminidase (BmFDL) for complex-type N-linked glycan synthesis in cultured silkworm cells. Biotechnol Lett 35:1009–1016
O'Reilly DR, Miller LK, Luckow VA (1992) Baculovirus expression vectors. In: Freeman WH (ed) A laboratory manual. Oxford University Press, New York, pp 139–166
Shih JD, Fitzgerald MC, Sutherlin M, Hunter CP (2009) The SID-1 double-stranded RNA transporter is not selective for dsRNA length. RNA 15:384–390
Soejima Y, Lee JM, Nagata Y, Mon H, Iiyama K, Kitano H, Matsuyama M, Kusakabe T (2013) Comparison of signal peptides for efficient protein secretion in the baculovirus-silkworm system. Cent Eur J Biol 8:1–7
Suganuma I, Ushiyama T, Yamada H, Iwamoto A, Kobayashi M, Ikeda M (2011) Cloning and characterization of a dronc homologue in the silkworm, Bombyx mori. Insect Biochem Mol Biol 41:909–921
Terenius O, Papanicolaou A, Garbutt JS, Eleftherianos I, Huvenne H, Kanginakudru S, Albrechtsen M, An C, Aymeric J-L, Barthel A, Bebas P, Bitra K, Bravo A, Chevalier F, Collinge DP, Crava CM, de Maagd RA, Duvic B, Erlandson M, Faye I, Felföldi G, Fujiwara H, Futahashi R, Gandhe AS, Gatehouse HS, Gatehouse LN, Giebultowicz JM, Gómez I, Grimmelikhuijzen CJP, Groot AT, Hauser F, Heckel DG, Hegedus DD, Hrycaj S, Huang L, Hull JJ, Iatrou K, Iga M, Kanost MR, Kotwica J, Li C, Li J, Liu J, Lundmark M, Matsumoto S, Meyering-Vos M, Millichap PJ, Monteiro A, Mrinal N, Niimi T, Nowara D, Ohnishi A, Oostra V, Ozaki K, Papakonstantinou M, Popadic A, Rajam MV, Saenko S, Simpson RM, Soberón M, Strand MR, Tomita S, Toprak U, Wang P, Wee CW, Whyard S, Zhang W, Nagaraju J, Ffrench-Constant RH, Herrero S, Gordon K, Swevers L, Smagghe G (2011) RNA interference in Lepidoptera: an overview of successful and unsuccessful studies and implications for experimental design. J Insect Physiol 57:231–245
The International Silkworm Genome Consortium (2008) The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol 38:1036–1045
Tsang SY, Moore JC, Huizen RV, Chan CWY, Li RA (2007) Ectopic expression of systemic RNA interference defective protein in embryonic stem cells. Biochem Biophys Res Commun 357:480–486
Watanabe S, Kokuho T, Takahashi H, Takahashi M, Kubota T, Inumaru S (2002) Sialylation of N-glycans on the recombinant proteins expressed by a baculovirus-insect cell system under beta-N-acetylglucosaminidase inhibition. J Biol Chem 277:5090–5093
Xu J, Nagata Y, Mon H, Li Z, Zhu L, Iiyama K, Kusakabe T, Lee JM (2013) Soaking RNAi-mediated modification of Sf9 cells for baculovirus expression system by ectopic expression of Caenorhabditis elegans SID-1. Appl Microbiol Biotechnol 97:5921–5931
Xue J, Qiao N, ZHANG W, Cheng RL, Zhang XQ, Bao YY, Xu YP, Gu LZ, Han JDJ, Zhang CX (2012) Dynamic interactions between Bombyx mori nucleopolyhedrovirus and its host cells revealed by transcriptome analysis. J Virol 86:7345–7359
Zhang JY, Pan MH, Sun ZY, Huang SJ, Yu ZS, Liu D, Zhao DH, Lu C (2010) The genomic underpinnings of apoptosis in the silkworm, Bombyx mori. BMC Genomics 11:611
Acknowledgments
This work was supported in part by grants KAKENHI nos. 22248003, 22248004, and 23580077 from the Japan Society for the Promotion of Science. The cost of publication was supported in part by the Research Grant for Young Investigators of Faculty of Agriculture, Kyushu University.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 147 kb)
Rights and permissions
About this article
Cite this article
Xu, J., Mon, H., Kusakabe, T. et al. Establishment of a soaking RNA interference and Bombyx mori nucleopolyhedrovirus (BmNPV)-hypersensitive cell line using Bme21 cell. Appl Microbiol Biotechnol 97, 10435–10444 (2013). https://doi.org/10.1007/s00253-013-5279-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5279-x