
Abstract
RNA interference (RNAi) has become an efficient tool for inducing resistance to viruses in many organisms. In this study, Escherichia coli cells were engineered to produce stable double-stranded RNA (dsRNA) against the nucleopolyhedrosis virus to elicit RNAi in silkworms. The immediate-early-1 (ie-1) and late expression factor-1 (lef-1) genes of the Bombyx mori nucleopolyhedrovirus (BmNPV) involved in viral DNA multiplication were cloned in the plasmid L4440 under the influence of the double T7 promoter and transformed to E. coli HT115 DE3 host cells. On induction with isopropyl β-d-thiogalactopyranoside, these cells efficiently produced dsRNA of the cloned genes. The B. mori larvae were fed with 50 µL of E. coli cells expressing ie-1 and lef-1 dsRNAs (each approximately 25 µg) to elicit RNAi. The semi-quantitative and quantitative PCR analysis of RNA from the midgut of the dsRNA-fed larvae revealed a significant reduction in the expression of the target genes involved in BmNPV multiplication, which restricted virus copy numbers to 100 compared with 1.9 × 105 in the infected controls. Furthermore, the dsRNA-fed infected larvae showed > 50% increased survivability compared with the infected controls. The study revealed the successful use of bacteria as vectors for efficiently delivering dsRNA to elicit RNAi against BmNPV in silkworms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
In silkworms, Grasserie disease caused by the nucleopolyhedrovirus (NPV) is considered a major viral disease responsible for 20–50% cocoon crop loss in the sericulture industry [1]. The Bombyx mori NPV (BmNPV) is a natural pathogen of the mulberry silkworm and is of great economic concern to farmers in sericulture-practicing countries. Finding an effective treatment for controlling the virus has been difficult due to the sturdy nature of the virus. Grasserie outbreak is often observed in tropical sericultural countries that experience frequent fluctuations in humidity and temperature throughout the year, being one of the predisposing factors for infection. Because of the lack of preventive measures to stop the spread of the infection, burning of infected silkworms and performing meticulous disinfection prior to and also during rearing is the lone way out.
RNAi in Lepidoptera was first reported in 2001 at the Fifth International Workshop on Molecular Biology and Genetics of Lepidoptera, thereby generating extensive interest in the possibility of utilizing this technology to investigate gene function in Lepidoptera [2]. Successful RNAi experiments in B. mori were conducted to study the physiology related to embryonic development, postembryonic development, cocoon color formation, pigmentation, and ecdysis [3,4,5,6,7]. However, much later, RNAi was shown to be a useful tool for inducing resistance to viruses in organisms, particularly invertebrates lacking an adaptive immune system [8]. Based on these research findings, transgenic silkworms expressing hairpin RNAs against the BmNPV ie-1 gene were successfully developed [9,10,11]. A moderate inhibition of virus replication was also observed in silkworms expressing BmNPV lef-1 double-stranded RNA (dsRNA) by combining RNAi and B. mori germline transformation. Similar experiments were conducted to engineer silkworms through multigene RNAi against BmNPV infection in transgenic lines [12, 13].
Many in vitro and in vivo studies have reported the effective use of RNAi technology for viral gene silencing through B. mori germline transformation. However, the present study is the first report on employing non-pathogenic bacteria as an oral feed/vector for delivering dsRNA to elicit RNAi against NPV genes in the silkworm B. mori. Using attenuated non-pathogenic bacteria as safe, effective, and inexpensive vectors for delivering dsRNA to elicit RNAi in target cells is the main emphasis of the present study. Bacteria-based RNAi is technically suitable for the production of large quantities of stable dsRNA and the development of environmentally safe pest control applications. Our study successfully showed that feeding of NPV-infected silkworms with bacterially produced dsRNA significantly decreased viral multiplication and increased the survivability of the silkworms.
Materials and methods
Collection of infected larvae and purification of BmNPV polyhedra
BmNPV-infected fifth instar silkworm larvae were collected from the rearing house. The haemolymph (white milky fluid) was collected by puncturing the prolegs of the infected larvae, transferred into sterile tubes containing few specks of phenylthiourea, and filtered through two layers of cheesecloth/non-absorbent cotton pad. The polyhedra were pelleted through centrifugation at 250 × g for 10 min at 4 °C, and the pellet was suspended in distilled water, followed by washing for 3 times through centrifugation at 250 × g for 10 min at 4 °C. The pellet was further washed three times with 1 M NaCl to obtain a white pellet of pure polyhedra adhering to the walls of the tubes. The pellet of pure polyhedra was suspended in distilled water and stored at 4 °C for use as per requirement.
Viral DNA extraction, PCR amplification, and sequencing
The viral DNA was isolated from the purified occlusion bodies (OBs) by using 2PK buffer containing 200 mM Tris, 25 mM ethylenediaminetetraacetic acid (EDTA), 300 mM NaCl, and 2% sodium dodecyl sulphate, followed by proteinase K treatment for 30 min. The DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1), precipitated in 2.5 volumes of ethanol, washed with 70% ethanol, and suspended in 10 mM Tris–1 mM EDTA. 35 ng of viral DNA was used to amplify the 310-bp product of the immediate-early-1 (ie-1) (NCBI Accession Number L33180.1) and late expression factor (lef-1) (NCBI Accession Number L33180.1) genes [12] using specific primers (Table 1). PCR amplification was performed using the following conditions: initial denaturation of 5 min at 94 °C; 30 cycles of 30 s at 94 °C, 30 s at 52 °C, and 1 min at 72 °C; and a final extension of 10 min at 72 °C. The PCR products were separated on 1.8% agarose gel using a 100-bp DNA ladder. 50 ng of the PCR product was used for DNA sequencing. The sequences obtained were verified in the NCBI database using the BLASTN search algorithm. To determine the virus copy number at different time points, quantitative PCR was performed using genomic DNA isolated from the BmNPV-infected larval midgut samples collected at different time points from 6 to 72 hours post infection (hpi), and the gp41 copy number was determined.
Construction of plasmid expressing ie-1 and lef-1 dsRNAs and its induction in the bacterial expression system
The viral genes ie-1 and lef-1 were PCR amplified using BmNPV viral DNA as a template under the following conditions: initial denaturation of 3 min at 95 °C; 30 cycles of 1 min at 95 °C, 30 s at 58 °C, and 1 min at 72 °C; and final extension at 72 °C for 8 min. The PCR products were separated on 1.8% agarose gels, visualised through ethidium bromide staining, and confirmed by sequencing. The PCR products were then cloned individually into the pJet 1.2 vector, and the positive clones were restriction digested using XhoI and XbaI to release a 310-bp insert. This insert was subsequently subcloned into L4440, a commercially available vector that has an Escherichia coli origin of replication, two T7 promoters in an inverted orientation flanking the multiple cloning sites (MCSs) for generating dsRNA from the cloned insert, and an ampicillin resistance gene for selection The resulting clones, namely L4440-ie-1, L4440-lef-1, and L4440 without any insert (plain vector), were transformed into competent E. coli HT115 (DE3) cells possessing tetracycline resistance as a selectable marker and lacking RNase III activity for inducing dsRNA production.
The transformed colonies were grown at 37 °C in a shaker incubator until the cells attained OD600 of 0.4. Then, the cells were induced with 0.25 mM IPTG, allowed to grow for an additional 4 h at 37 °C, and harvested through centrifugation at 8000 rpm to isolate dsRNA. The cDNA was synthesised from the isolated dsRNA using the PrimeScript cDNA synthesis kit (DSS Takara Bio Ltd.), and the presence of the cloned genes as well as induction of lef-1 dsRNA, ie-1 dsRNA, and L4440 were confirmed through cDNA amplification using gene-specific primers.
Inoculation of virus for bioassay experiments
The uncontaminated OBs of BmNPV isolated from the haemolymph of the infected larvae (CSR2: Silkworm strain) were used for infecting freshly moulted individual fifth instar larvae by feeding mulberry leaves coated with 10 µL suspension of 2000 OBs/mL [1]. Viral multiplication at different time points from 6 to 72 hpi was quantitatively analysed using gp41 primers.
Insect rearing and feeding bioassays
Large-scale production of dsRNA for bioassay experiments was performed by inoculating 1% of the overnight grown E. coli HT115 DE3 cells transformed with ie-1 and lef-1 clones and the L4440 vector into 50 mL Luria–Bertani (LB, Himedia Laboratories Pvt Ltd.) broth and incubated at 37 °C in a shaker incubator. The cells were induced with 0.25 mM IPTG when the cultures attained 0.4 OD and were allowed to grow further. After 4 h of growth, the complete 50 mL bacterial cells were harvested and suspended in 1 mL autoclaved water to obtain a concentration of 50 × for feeding silkworms.
Approximately 250 newly moulted 5th instar healthy silkworm larvae were collected from the rearing house and divided into 5 groups, with each containing 50 larvae. Except the first group, all larvae from the remaining four groups were inoculated individually per os via ingestion of a 3-cm2 piece of mulberry leaf coated with 10 µL suspension of 2000 OBs of BmNPV and 50 µL bacterial cells (50×) containing approximately 25 µg of dsRNA (ingested doses of virus and bacteria are an average calculation). The first group was maintained as an uninfected control, that is, healthy larvae without any infection or dsRNA, the second group included larvae inoculated with induced L4440 cells, the third group included larvae inoculated with induced ie-1 dsRNA bacterial cells, the fourth group included larvae inoculated with induced lef-1 dsRNA bacterial cells, and the fifth group of larvae was inoculated with 2000 OBs/larva and maintained as the infected control. After the inoculated larvae consumed the entire dosage of bacterial cells, they were further reared on fresh mulberry leaves along with uninfected controls as per the feeding, cleaning, and sanitation schedules until the onset of spinning of cocoons (approximately 5–6 days). However, for molecular analysis, the midgut tissue from an aliquot of larvae from each of the treatments was dissected at 24 hpi. The tissue was rinsed/cleansed in autoclaved water to clear off the digested leaf materials and used for isolating genomic DNA as well as total RNA for the analysis of viral multiplication and viral gene expression. The number of dead larvae against the number of healthy larvae (control) in each group was recorded to determine the survival percentage in each treatment group until the onset of spinning of cocoons.
Quantitative analysis of viral copy number and differential viral gene expression in dsRNA-fed silkworms and control silkworms
To analyse viral multiplication, quantitative PCR was performed using genomic DNA isolated from the larval midgut to compare the gp41 gene copy number in the midgut of the dsRNA-fed silkworms with that in the control samples. The PCR conditions included an initial denaturation of 5 min at 94 °C; 30 cycles of 30 s at 94 °C, 30 s at 55 °C, and 1 min at 72 °C; and a final extension of 10 min at 72 °C. To analyse the differential gene expression, total RNA was extracted from the experimental (dsRNA-fed) as well as control midgut samples of B. mori by using RNA isoplus reagent (DSS Takara Bio Ltd.) according to the manufacturer’s instructions. Each RNA sample (0.5–1 µg) was used for cDNA synthesis in a 20-µL reaction mixture under the following reaction conditions: 37 °C for 10 min, 42 °C for 1 h, 75 °C for 10 min, and 5 °C for 5 min. Semi-quantitative PCR was performed with 2 µL of cDNA as a template in 20-µL reaction mixtures using gene-specific primers. The PCR programme included an initial denaturation of 5 min at 94 °C; 30 cycles of 30 s at 94 °C, 30 s at 58 °C, and 1 min at 72 °C; and a final extension of 10 min at 72 °C. The PCR products were analysed through 1.8% agarose gel electrophoresis along with a 100-bp marker. Based on the semi-quantitative data, quantitative or real-time PCR analysis was performed using gene-specific primers to determine the differential expression of viral genes in the control and treatment groups. Using SYBR Premix Ex-TaqII (DSS Takara Bio Ltd.), real-time PCR was performed under the following reaction conditions: initial denaturation of 5 min at 94 °C; 30 cycles of 30 s at 94 °C, 30 s at 52 °C, and 1 min at 72 °C in triplicate. The relative fold change in gene expression was calculated; the housekeeping gene β-actin was used as an internal control to normalise the reactions.
Statistical analysis
Data are presented as mean ± standard deviation of three independent experiments with 50 larvae in each experiment. Statistical analysis was performed using ANOVA, followed by Dunnett’s post hoc test (http://www.graphpad.com/quickcalcs/) to determine whether significant differences existed between the control and dsRNA-treated samples. P = 0.0002 and < 0.0001 were considered as significant (represented by asterisks *** and ****, respectively).
Results
BmNPV infection and isolation of essential viral gene(s) involved in BmNPV multiplication
Symptoms of the larvae with Grasserie disease were observed from 6 hpi and at 24 hpi. The larvae became restless and stopped feeding due to the severity of BmNPV infection. The larvae appeared with a shiny white translucent skin, and swollen intersegmental membrane and body segments. At an advanced stage of infection, that is, 5–6 days after infection, the body wall ruptured and white turbid haemolymph containing a large number of OBs oozed out and turned blackish brown within 3–5 h due to oxidation (Fig. 1a). The haemolymph was collected to purify the OBs before they became oxidised. Genomic DNA was isolated from the purified polyhedra and yielded a 310-bp amplicon upon amplification with BmNPV-specific ie-1 and lef-1 primers (Fig. 1b), and sequencing of the ie-1 and lef-1 PCR products confirmed that the amplified product was of BmNPV origin. No differences were observed at the nucleotide sequence level in the isolated ie-1 and lef-1 genes when verified against the NCBI Accession Number L33180.1. The viral copy numbers at different time points were determined by quantitative PCR, and in the midgut samples, < 100 copies of gp41 were present as early as 6 hpi. As time advanced, a substantial increase in copy numbers was observed, with the number becoming as high as 2.8 × 105 at 72 hpi. Melting curve analysis confirmed the gene-specific amplification using gp41 primer. gp41 is an important gene responsible for virion assembly and morphogenesis. In the early stage of BmNPV infection at 6 hpi, viral proliferation was detected in the larval midgut with a relative copy number of 21.2 copies. As the infection progressed, the viral proliferation levels increased from approximately 21.2 copies to 2.8 × 105 copies within 6–72 hpi (Fig. 1c).

Analysis of viral genes in silkworms infected with Bombyx mori nucleopolyhedrovirus (BmNPV). a Fifth instar BmNPV-infected silkworms with swollen integuments, putrefied BmNPV-infected larvae (red arrow: white milky fluid turned brown due to oxidation). b Viral genes ie-1 and lef-1 amplified from the genomic DNA isolated from the infected silkworms for cloning into the dsRNA-producing L4440 vector. c Quantitative PCR for determining the virus copy number at different time intervals. (Color figure online)
Cloning and expression analysis of ie-1 and lef-1 dsRNAs in bacteria
The L4440 vector consists of a double T7 promoter in an inverted orientation flanking the MCSs, and the ie-1 and lef-1 genes of 310 bp were cloned in between these T7 promoters at XhoI and XbaI sites to generate respective dsRNAs (ie-1 and lef-1 genes were cloned as different constructs) (Fig. 2a). After transformation into bacteria, few selected colonies were subjected to plasmid isolation, followed by restriction digestion yielded an insert of size 310 bp for XhoI and XbaI restriction enzymes, respectively (Fig. 2b). The clones were also confirmed by PCR using gene- and vector-specific primers, and the confirmed clones were transformed into HT115 DE3 cells induced with 0.25 mM IPTG. The induction of dsRNA was confirmed by isolating total RNA and preparation of cDNA from the induced L4440 vector alone, L4440 vector containing lef-1, and L4440 vector containing ie-1 genes of interest. The semiquantitative expression analysis showed expected 310-bp amplification of lef-1 and ie-1 genes in the induced L4440-lef-1 and L4440-ie-1 clones, whereas no such amplification was observed in the induced L4440 vector alone. This clearly indicated the expression of dsRNA from the cloned lef-1 and ie-1 genes of BmNPV in bacteria (Fig. 2c). To separately determine the effect of ie-1 and lef-1 dsRNAs, both the ie-1 and lef-1 genes were cloned separately as two different constructs. The induced bacterial cells (dsRNA) were fed orally to silkworms through mulberry leaves along with the virus inoculum.

Cloning and expression analysis of dsRNA produced in bacteria. a L4440 vector map:addgene. b Restriction digestion of ie-1 and lef-1 clones using XhoI and XbaI enzymes releasing the 310-bp insert (1: uncut plasmid, 2–3 ie-1 clones, 4–5 lef-1 clones, M marker). c Semiquantitative analysis to confirm the induction of dsRNA in bacteria before feeding to silkworms
Effect of ie-1 and lef-1 dsRNAs on genes involved in viral multiplication in BmNPV-infected silkworms
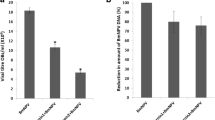
The RNAi effect on NPV multiplication in the dsRNA-fed silkworms was analysed on the basis of the gp41 gene copy number in the midgut tissue dissected after 24 hpi. The copy number analysis showed < 110 and 118 copies of the gp41 gene in the ie-1 dsRNA- and lef-1 dsRNA-fed silkworms, respectively. In the NPV-infected and NPV + L4440-fed silkworms, the relative copy numbers of the gp41 gene were 1.9 × 105 and 1.8 × 105, respectively (Fig. 3a). This clearly indicated a significant (P < 0.0001) decrease in the gp41 gene copy number in the dsRNA-fed silkworms. Furthermore, quantitative PCR analysis was used to compare the differential expression of the viral genes ie-1, lef-1, and gp41 in the dsRNA-fed infected and NPV-infected silkworms. The quantitative PCR analysis showed a significant (P < 0.0001) decrease in the transcript levels of gp41, ie-1, and lef-1 genes in the ie-1 dsRNA- and lef-1 dsRNA-fed infected silkworms compared with the controls. The ie-1 dsRNA-fed silkworms showed a 6-fold decrease in the expression of all three viral genes and the lef-1 dsRNA-fed silkworms showed a 5-fold decrease in ie-1 gene expression and a 6-fold decrease in lef-1 and gp41 gene expression (Fig. 3b). This result indicated the effect of the core small-interfering RNA pathway on the RNAi machinery that affected viral copy numbers, followed by effective gene silencing with both the constructs fed to silkworms despite a slight variation in fold change in viral gene expression between the constructs.

Expression analysis of genes involved in NPV multiplication. The fifth instar larvae were collected after being fed with ie-1 dsRNA- and lef-1 dsRNA-expressing bacteria. Total RNA was isolated followed by cDNA synthesis, and gene expression was analysed using quantitative PCR. a Copy number estimation using gp41-specific primers in bioassay samples dissected from NPV-infected and dsRNA-fed infected silkworms. b dsRNA-feeding bioassay: quantitative PCR analysis to compare the expression levels of the viral genes gp41, ie-1, and lef-1 in infected silkworms fed with dsRNAs. The gene expression values were normalised with actin primers. The data represent the mean of three independent experiments. Values were compared using the ANOVA test. *** and ****Significant difference at P = 0.0002 and < 0.0001, respectively, compared with the controls
Survivability analysis of ie-1 dsRNA- and lef-1 dsRNA-fed NPV-infected silkworms
To determine the effect of dsRNA on the infected larvae, the number of dead larvae against the number of larvae that survived after BmNPV infection until the onset of spinning of cocoons was calculated for different treatments, that is, ie-1 and lef-1 dsRNA treatments. In the larvae fed with NPV and the NPV + L4440 vector alone, 1–2% of the larvae looked healthy and survived till cocoon formation, whereas all the remaining larvae showed disease symptoms. The infected silkworms fed with dsRNA constructs independently, that is, ie-1 dsRNA-fed BmNPV-infected larvae and lef-1 dsRNA-fed BmNPV-infected larvae, showed 52.8% and 58.8% survival, respectively. Thus, both the dsRNA constructs were effective in supressing viral multiplication. The results indicated > 50% increase in the survivability of virus-infected silkworms fed with lef-1 and ie-1 dsRNAs compared with the control infected silkworms. Although a differential expression of viral genes was observed at the molecular level, not much variation was observed in the survivability and both the constructs were able to elicit RNAi against the viral genes (Fig. 4a, b).

Bioassay experiments to validate the effect of lef-1 and ie-1 dsRNAs in BmNPV-infected silkworms. a Photographs of rearing trays of different treatments consisting of silkworms fed with dsRNAs and controls (red circle indicates infected/dead larvae). b Graphical representation of feeding bioassay results showing survivability of ie-1 dsRNA- and lef-1 dsRNA-fed NPV-infected silkworms in comparison with only 2–3% survivability of the larvae fed with NPV and the NPV + L4440 vector alone, whereas controls (uninfected) showed no disease symptoms. The data represent the mean of three independent experiments with 50 larvae per experiment. Values were compared using the ANOVA test. ****Results that are significant at P < 0.0001 between the treatment and control groups. (Color figure online)
Discussion
Targeting ie-1 and lef-1 for cloning and expression analysis of ie-1 and lef-1 dsRNAs in bacteria
The dsRNA produced makes use of the bacterial system for producing dsRNA against BmNPV viral genes. Therefore, we developed a dsRNA construct using the L4440 vector that has two T7 promoters in an inverted orientation flanking the MCSs. The virus-specific gene was cloned in between the T7 promoters flanking the MCSs to generate dsRNA. Two important genes involved in viral DNA replication were targeted, namely, ie-1 (immediate-early-1) that oligomerises within the cytosol and gets involved with the viral DNA replication machinery [9, 14], and lef-1 (late expression factor), a DNA primase of the replication complex responsible for viral DNA replication [13,14,15]. Both the genes are expressed between 6 and 12 hpi, and expression of other late and very late genes are dependent on the onset of the expression of these genes. In other similar studies in vitro, high levels of silencing of the target gene was described wherein transgenic B. mori silkworms expressing hairpin RNAs against BmNPV genes were successfully developed [9,10,11,12,13]. Thus, both the genes are appropriate targets for RNAi-mediated gene silencing. In our study, the ie-1 and lef-1 genes of size 310 bp were cloned separately as two different constructs in the L4440 vector. Both the clones were confirmed by restriction digestion to release an insert of 310 bp, and the clones were also sequenced for the confirmation of respective genes. Both the dsRNAs induced by IPTG were confirmed by PCR using gene-specific primers and fed to the infected silkworms in the form of transformed bacterial cells to study the effect of the dsRNAs in the silkworms (Fig. 2c). The induced bacterial cells (dsRNA) were orally fed to silkworms through mulberry leaves along with the virus, and after 24 h of feeding, the midgut tissue was dissected for molecular analysis.
ie-1 dsRNA- and lef-1 dsRNA-fed silkworms express lower copy numbers of gp41 and NPV-associated genes
The quantitative PCR analysis was performed to compare the differential expression of the viral genes ie-1, lef-1, and gp41 in the dsRNA-fed infected silkworms and the control group. The NPV-infected silkworms fed with ie-1 and lef-1 dsRNA constructs showed approximately 5- to 6-fold decrease in the transcript levels of gp41, which is an important gene responsible for virion assembly. The expression levels of gp41, ie-1, and lef-1 in the lef-1 dsRNA-fed silkworms were considerably lower than those in the ie-1 dsRNA-fed silkworms. The variations found in the expression levels of ie-1 and lef-1 transcripts in the ie-1 dsRNA- and lef-1 dsRNA-fed silkworms could probably be due to the fact that lef-1 transcript initiation by the ie-1 product is temporal, that is, the lef-1 transcript level is influenced by ie-1 directly or indirectly [13]; early genes such as ie-1 are generally encoded proteins of regulatory functions and modifications of the host process, and generally, early gene transcription takes place within 0–6 hpi, whereas late and very late transcription is dependent on early viral gene expression and viral DNA replication [16, 17] (Fig. 3b). Between both the constructs that were fed independently to the infected silkworms, the lef-1 dsRNA construct was considerably more effective than the ie-1 dsRNA construct, and the mRNA levels of gp41 and ie-1 genes were also significantly lower in the lef-1 dsRNA construct than in the ie-1 dsRNA construct. But it was important to note that there was no significant difference in the transcript levels of viral genes between the two dsRNA constructs, as both the constructs showed decreased viral transcripts. The decrease in the copy number of gp41 in ie-1 and lef-1 dsRNA fed infected silkworms when compared to infected control can be attributed to the fact that gp41 gene is one of the of the baculovirus core genes that codes the O-glycosylated protein of the occlusion-derived virion phenotype and plays a very crucial role in virion morphogenesis. The decrease in the expression level of ie-1 and lef-1 led to a decrease in the expression of gp41, which plays a crucial role in budded virion formation and executes functions in baculovirus virion morphogenesis and assembly [18]. In this study, as the target was early genes, the decrease in ie-1 and lef-1 expression due to RNAi led to decreased viral DNA multiplication, followed by a decrease in the expression of other genes responsible for virus proliferation, finally affecting virion formation. This clearly indicated that if gene expression is silenced at an early stage, that is, targeting early genes will help in the effective arrest of viral proliferation and corresponding increase in the survival rate of the infected silkworms. Thus, the study showed that dsRNA-fed silkworms increased the tolerance level to BmNPV infection by producing viral mRNA-specific siRNA. Studies investigating the antiviral defence mechanism in honey bees also showed that in addition to inducing RNAi, dsRNA is engaged in a previously uncharacterised non-sequence-specific immune pathway/the involvement of innate immune pathways (i.e., Jak-Stat, Toll, and Imd) and non-sequence-specific dsRNA-mediated immune responses [19].
Feeding dsRNA orally for effective delivery of dsRNA into the host silkworm B. mori
The efficacy of RNAi depends both on the biology of the organism and the method of dsRNA delivery, that is, intrinsic (efficiency of dsRNA uptake, relative turnover of target mRNA/protein, signal amplification, and systemic spread) and extrinsic factors (concentration, target gene selection, transcript localisation, dsRNA synthesis, and route of introduction). Lack of a stable and effective dsRNA delivery system is one of the major drawbacks associated with transgenic RNAi technology. Therefore, most current studies aim at improving existing methodologies and adopting innovative technologies to increase the efficiency of RNAi. Many studies have exploited alternative routes for dsRNA delivery [20]. Studies conducted by Timmons and Fire have shown that systemic gene silencing could be attained in the nematode Caenorhabditis elegans when it ingested E. coli engineered to produce interfering RNAs. This discovery established an additional method to introduce dsRNA into organisms for triggering RNAi [21,22,23,24,25]. Among all the existing methods suggested for the delivery of dsRNA into the host, oral delivery/ingestion is a less invasive and potentially high-throughput method for delivering dsRNA [26]. The overall study suggests that feeding of bacterially produced dsRNA at fifth instar to silkworms was effective in silencing the genes expressed during BmNPV infection (Fig. 3a). Bacterially expressed dsRNA when fed to the insect larvae produced effects similar to those produced by other widely used techniques. As the dsRNA is expressed in the bacteria, degradation is also prevented to a great extent. In our study, we observed that the dsRNA was stable for 24 h both in the haemolymph and midgut, but slowly degraded at 36–48 h (data shown in Supplementary File). Moreover, it was observed that feeding dsRNA is better in terms of specificity and reduced chances of secondary siRNA production in the target organism, which may lead to off-target effects [27,28,29,30]. This study showed that feeding bacterially expressed dsRNA led to a significant drop in the transcript levels of viral genes and restricted viral multiplication in the host compared with that in the control. In the RT-PCR analysis, larvae fed with ie-1 and lef-1 dsRNAs showed a substantial reduction in the expression of target genes involved in BmNPV multiplication and significantly increased the survivability of the infected silkworms (Fig. 4b). Oral delivery of dsRNA is a high-throughput method for RNAi delivery in insects intolerant to injection and for large-scale field applications for RNAi-mediated pest control. Hence, bacteria expressing the desired dsRNA(s) against viral gene(s) in insects may be effectively used for combating viral infections in the silkworm B. mori. The results obtained indicate the successful use of non-pathogenic bacteria as a vector for delivering dsRNA and eliciting RNAi against BmNPV in silkworms. Although the survival of larvae was less, the efficacy of dsRNA can be improved by feeding multiple doses of dsRNA [25] or by feeding both dsRNAs (i.e., ie-1 and lef-1 dsRNAs) together. This study showed that oral feeding of dsRNA produced in bacteria is one of the reliable method for introducing dsRNA into silkworms, and > 50% survival of the infected silkworms was achieved by this method. Therefore, this technology will be useful in minimising the cocoon crop loss to a greater extent.
Conclusion
This technology provides a compatible method to achieve RNAi for various applications due to proven clinical safety of non-pathogenic bacteria as a gene carrier. Because dsRNA is produced inside the bacteria, the whole process becomes stable and cost-effective for mass multiplication. This technology is highly regarded as potentially therapeutic due to its high efficacy, low toxicity, and ability to allow the establishment of a stable gene silencing system in the laboratory against NPV genes that activate viral multiplication.
References
Khurad AM, Mahulikar A, Rathod MK, Manoj R, Kanginakudru S, Nagaraju J (2004) Vertical transmission of nucleopolyhedrovirus in the silkworm, Bombyx mori L. J Invertebr Pathol 87:8–15
Terenius O, Papanicolaou A, Garbutt JS, Eleftherianos I, Huvenne H et al (2011) RNA interference in Lepidoptera: an overview of successful and unsuccessful studies and implications for experimental design. J Insect Physiol 57:231–245
Tabunoki HS, Ninagi O, Fujii H, Banno Y, Nozaki MA (2004) Carotenoid-binding protein (CBP) plays a crucial role in cocoon pigmentation of silkworm (Bombyx mori) larvae. FEBS Lett 567:175–178
Ohnishi A, Hull JJ, Matsumoto S (2006) Targeted disruption of genes in the Bombyx mori sex pheromone biosynthetic pathway. Proc Natl Acad Sci USA 103:4398–4403
Liu W, Yang F, Jia S, Miao X, Huang Y (2008) Cloning and characterization of Bmrunt from the silkworm Bombyx mori during embryonic development. Arch Insect Biochem Physiol 69:47–59
Dai H, Ma L, Wang J, Jiang R, Wang Z, Fei J (2008) Knockdown of ecdysis-triggering hormone gene with abinary UAS/GAL4 RNA interference system leads to lethal ecdysis deficiency in silkworm. Acta Biochim Biophys Sin 40:790–795
Masumoto M, Yaginuma T, Niimi T (2009) Functional analysis of Ultrabithorax in the silkworm, Bombyx mori using RNAi. Dev Genes Evol 219:437–444
La Fauce K, Owens L (2012) RNA interference with special reference to combating viruses of crustacea. Indian J Virol 23:226–243
Kanginakudru S, Royer C, Edupalli SV, Jalabert A, Mauchamp B, Chandrashekaraiah, Prasad SV, Chavancy G, Couble P, Nagaraju J (2009) Targeting ie-1 gene by RNAi induces baculoviral resistance in Lepidopteran cell lines and in transgenic silkworms. Insect Mol Biol 16:635–644
Tamura T, Thibert C, Royer C, Kanda T, Abraham E, Kamba M et al (2000) Germline transformation of the silkworm Bombyx mori L. using a piggyBac transposon-derived vector. Nat Biotechnol 18:81–84
Isobe R, Kojima K, Matsuyama T, Quan GX, Kanda T, Tamura T, Sahara K, Asano SI, Bando H (2004) Use of RNAi technology to confer enhanced resistance to BmNPV on transgenic silkworms. Arch Virol 149:1931–1940
Subbaiah EV, Royer C, Kanginakudru S, Satyavathi VV, Babu AS, Sivaprasad V, Chavancy G, Darocha M, Jalabert A, Mauchamp B, Basha I, Couble P, Nagaraju J (2013) Engineering silkworms for resistance to Baculovirus through multigene RNA interference. Genetics 193:63–75
Zhang P, Wang J, Lu Y, Hu Y, Xue R, Cao G, Gong C (2014) Resistance of transgenic silkworm to BmNPV could be improved by silencing ie-1 and lef-1 genes. Gene Ther 21:81–88
Olson VA, Wetter JA, Friesen PD (2002) Baculovirus transregulator IE1 requires a dimeric nuclear localization element for nuclear import and promoter activation. J Virol 76:9505–9515
Mikhailov VS, Rohrmann GF (2002) Baculovirus replication factor LEF-1 is a DNA primase. J Virol 76:2287–2297
Acharya A, Sriram S, Sehrawat S, Rahman M, Sehgal D, Gopinathan KP (2002) Bombyx mori nucleopolyhedrovirus: molecular biology and biotechnological applications for large scale synthesis of recombinant protein. Curr Sci 83:4
Katsuma S, Mita K, Shimada T (2007) ERK- and JNK-dependent signaling pathways contribute to Bombyx mori nucleopolyhedrovirus infection. J Virol 81:13700–13709
Li Y, Shen S, Hu L, Deng F, Vlak JM, Hu Z, Wang H, Wang M (2018) The functional oligomeric state of tegument protein GP41 is essential for Baculovirus budded virion and occlusion-derived virion assembly. J Virol 92:e02083-17
Brutscher LM, Flenniken ML (2015) RNAi and antiviral defense in the honey bee. J Immunol Res. https://doi.org/10.1155/2015/941897
Scott JG, Michel K, Bartholomay LC, Siegfried BD, Hunter G, Smagghe WB, Zhu KY, Douglas AE (2013) Towards the elements of successful insect RNAi—review. J Insect Physiol 59:1212–1221
Turner CT, Davy MW, MacDiarmid RM, Plummer KM, Birch NP, Newcomb RD (2006) RNA interference in the light brown apple moth, Epiphyas postvittana (Walker) induced by double-stranded RNA feeding. Insect Mol Biol 15:383–391
Baum JA, Bogaert T, Clinton W, Heck GR, Feldmann P, Ilagan O, Johnson S, Plaetinck G, Munyikwa T, Pleau M, Vaughn T, Roberts J (2007) Control of coleopteran insect pests through RNA interference. Nat Biotechnol 25:1322–1326
Tian H, Peng H, Yao Q, Chen H, Xie Q, Tang B, Zhang W (2009) Developmental control of a Lepidopteran pest Spodoptera exigua by ingestion of bacterial expressing dsRNA of a non-midgut gene. PLoS ONE 4:e6225
Surakasi VP, Mohamed AM, Kim Y (2011) RNA interference of beta 1 integrin subunit impairs development and immune responses of the beet armyworm, Spodoptera exigua J. Insect Physiol 57:1537–1544
Yang J, Han Z (2014) Efficiency of different methods for dsRNA delivery in cotton bollworm (Helicoverpa armigera). J Integr Agric 13:115–123
Piot N, Snoeck S, Vanlede M, Smagghe G, Meeus I (2015) The effect of oral administration of dsRNA on viral replication and mortality in Bombus terrestris. Viruses 7:3172–3185
Timmon L, Fire A (1998) Specific interference by ingested dsRNA. Nature 395:854
Timmon L, Court DL, Fire A (2001) Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263:103–112
Yu N, Christiaens O, Liu J, Niu J, Cappelle K, Caccia S, Huvenne H, Smagghe G (2013) Delivery of dsRNA for RNAi in insects: an overview and future directions. Insect Sci 20:4–14
Yogindran S, Rajam MV (2015) RNA interference strategy for crop protection against insect pests. In: Soberon M, Gao Y, Bravo A (eds) Bt resistance, characterization and strategies for GM crops producing Bacillius thuringiensis Toxins. CABI Biotechnology Series 4. CAB International, Oxfordshire, pp 162e172
Acknowledgements
This Research was funded by Central Silk Board, Bengaluru and Technical Collaboration was provided by Professor M.V. Rajam, Department of Genetics, UDSC, New Delhi. The authors would like to acknowledge Dr. Geetha Murthy, Scientist, CSGRC, Hosur and Editingindia for meticulously editing the Manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ismail, S., Tulsi Naik, K.S., Rajam, M.V. et al. Targeting genes involved in nucleopolyhedrovirus DNA multiplication through RNA interference technology to induce resistance against the virus in silkworms. Mol Biol Rep 47, 5333–5342 (2020). https://doi.org/10.1007/s11033-020-05615-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05615-z