
Abstract
Investigating the environmental influence on the community composition and abundance of denitrifiers in marine sediment ecosystem is essential for understanding of the ecosystem-level controls on the biogeochemical process of denitrification. In the present study, nirK-harboring denitrifying communities in different mud deposit zones of eastern China marginal seas (ECMS) were investigated via clone library analysis. The abundance of three functional genes affiliated with denitrification (narG, nirK, nosZ) was assessed by fluorescent quantitative PCR. The nirK-harboring microbiota were dominated by a few operational taxonomic units (OTUs), which were widely distributed in different sites with each site harboring their unique phylotypes. The mean abundance of nirK was significantly higher than that of narG and nosZ genes, and the abundance of narG was higher than that of nosZ. The inconsistent abundance profile of different functional genes along the process of denitrification might indicate that nitrite reduction occurred independently of denitrification in the mud deposit zones of ECMS, and sedimentary denitrification was accomplished by cooperation of different denitrifying species rather than a single species. Such important information would be missed when targeting only a single denitrifying functional gene. Analysis of correlation between abundance ratios and environmental factors revealed that the response of denitrifiers to environmental factors was not invariable in different mud deposit zones. Our results suggested that a comprehensive analysis of different denitrifying functional genes may gain more information about the dynamics of denitrifying microbiota in marine sediments.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrogen (N), a limiting element for biological productivity, occupies a central role in ocean biogeochemistry and exerts a significant influence on cycling of many other elements, in particular carbon and phosphorus [1, 2]. Thus, the cycling of N is of great importance in maintaining ecosystem function in both terrestrial and aquatic systems [3]. Denitrification is one of the processes within the nitrogen cycle responsible for the removal of excess fixed nitrogen from natural ecosystems and agricultural fields, as well as during wastewater treatment [4]. The process is a series of reactions of dissimilatory nitrate reduction mediated by microorganisms under anoxic/suboxic conditions [5, 6]. In the process of denitrification, nitrate serves as an electron acceptor alternative to O2 and is reduced to N2 via nitrite, NO, and N2O with different enzymes catalyzing different steps [6], i.e., NO3 − → NO2 −, nitrate reductase (narG) [7]; NO2 − → NO, either cytochrome cd1 (nirS) or Cu-containing (nirK) nitrite reductase, depending on the organism [8]; NO → N2O, cytochrome bc-containing nitric oxide reductase (norB); and N2O → N2, Cu-containing nitrous oxide reductase (nosZ) [9]. The genes encoding these reductases are generally used as biological makers for different reactions of the denitrification process [10].
Distinct from anammox, another nitrogen removal process, the process of denitrification can involve the release of N2O as a consequence of incomplete denitrification. N2O is a greenhouse gas and accounts for 7.9% (CO2 equivalent emission) of anthropogenic greenhouse gas emission [8]. Therefore, denitrification has attracted much attention in the agricultural and industrial fields [4]. Due to its importance, the biogeochemical dynamics of denitrification has been evaluated in a variety of environments, such as river and coastal sediments, agricultural zones, wetlands, and forest soil [3, 10–13]. In marine ecosystems, denitrifying microbes are classically involved in the decomposition and mineralization of dissolved and particulate organic nitrogen, which directly impacts nitrogen availability for primary producers. Marine sediments are important niches for nitrogen removal through denitrification [14], and the abundance and community composition of denitrifiers by measuring the denitrifying genes mentioned above in sediments are affected by many factors, including concentration of dissolved inorganic nitrogen (DIN) (NO3 −, NO2 −, NH4 +), organic matter, dissolved oxygen, and redox potential [10]. Investigating the environmental influence on the community composition and determining the key factors that limit and control the abundance of denitrifiers are essential for understanding the ecosystem-level controls on the biogeochemical process of denitrification.
The Bohai Sea, Yellow Sea, and East China Sea (ECS) are three coastal seas that constitute the eastern China marginal seas (ECMS) [15]. As an example of the river-dominated ocean margins, they receive a large input of sediments per year from the Yellow River and the Changjiang River [16], which together with sediments delivered by other rivers and the open ocean lead to the formation of several isolated mud zones [17, 18]. Different mud zones are unique in environmental conditions from one another due to varied sedimentary sources and ocean current disturbances. For example, the Changjiang Estuary mud zone and the Zhe-Min mud zone have higher sedimentation rates than the Cheju Island mud zone and South Yellow Sea mud zone, which are farther from land. Therefore, different mud zones could have enriched different microbial communities [19].
Since only 0.5 to 1% of total marine bacteria may be cultivated, the utilization of universal primers in polymerase chain reactions (PCR) to screen functional genes relevant to biogeochemical processes in natural environments increases extensively. The PCR-based approaches have revealed the presence of diverse denitrifier communities in marine ecosystems [20–25]. However, most previous studies have targeted a single functional gene, such as the genes of nitrate reductase (narG), nitrite reductase (nirS and nirK), nitric oxide reductase (norB), or nitrous oxide reductase (nosZ) [15, 16, 26]. In this study, we presented a detailed analysis of functional distribution of denitrifiers in the mud deposit zones of ECMS. The diversity, community composition, and spatial distribution of denitrifiers was explored with the nirK gene. The abundance of denitrifiers in different sediment layers was investigated by fluorescent quantitative PCR (qPCR) by using three functional genes that are narG, nirK, and nosZ. We believe that a survey with a combination of functional genes targeting different reactions would provide a more comprehensive insight into the denitrification process in coastal marine sediments.
Materials and Methods
Site Description
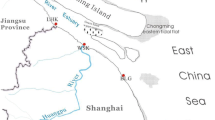
Five sampling sites which represent most of the typical sedimentary environments of ECMS were selected in this study (Fig. 1). The Changjiang Estuary mud zone (CEMZ), Zhe-Min mud zone (ZMMZ), Cheju Island mud zone (CIMZ), and South Yellow Sea mud zone (SYSMZ) are four primary mud deposit zones in ECMS which are formed by estuarine processes, tidal currents, and shelf circulation [17, 18]. ECS01 is located in CEMZ, where sediments are mainly imported from the Changjiang River and deposited under the effect of its diluted water, tidal actions, and coastal currents [17, 27]; sedimentation rates in this mud zone range from ∼2.0 to 6.0 cm/year [28]. ECS02 is located in ZMMZ, which is a mud wedge along the ECS inner shelf; sedimentation rates in this mud zone range from 0.4 to 1.2 cm/year [29]. Sediments of this zone stem from suspended sediments of the Changjiang River and resuspended sediments of CEMZ, which are transported by the southward Zhe-Min coastal current in winter when the strength of the northward Taiwan Warm Current declines [17]. ECS03 is located in the distal of CIMZ, and its sediments are derived from the mixed water from the Yellow Sea Warm Current, Taiwan Warm Current, and Changjiang River diluted water [30], with sedimentation rates ranging from 0.1 to 0.5 cm/year [31, 32]. SYS01 and SYS02 are located in SYSMZ, with sedimentation rates less than 0.1 cm/year [18, 31]. The muddy sediments in this area are mainly supplied by the Yellow Sea Warm Current.

Map showing locations of the five sampling sites in ECMS
Sample Collection and Environmental Factors
Mud deposit samples were collected from five stations of ECMS using a stainless steel box sampler during an open cruise of R/V Dong Fang Hong 2 in July, 2013. Two subcores were collected for molecular and physiochemical analysis, respectively. Pore waters at centimeter-scale resolution were obtained from the core for physiochemical analysis by using Rhizon samplers attached to vacuum test tubes; Rhizon samplers were inserted into sediments through pre-drilled holes in the core tube. The pore water samples were poisoned by HgCl2 and stored at 4 °C for laboratory analysis. Total organic carbon (TOC), total nitrogen (TN), stable carbon and nitrogen isotopes (δ13C and δ15N) in sediments, and dissolved inorganic nutrients (NO3 −, NO2 −, NH4 +, PO4 3−, and SiO3 2−) in pore waters were measured using a nutrient auto-analyzer (AA3; Seal Analytical Ltd., UK) as previously reported [19]. Salinity of the bottom water was recorded by a Conductivity-Temperature-Depth equipment (SBE 25 SEALOGGER; Sea-Bird Electronic Inc., USA) (Table S1).
DNA Extraction and nirK Gene Clone Library Analysis
Sediment (0–2 cm) DNA was extracted from replicate subcore samples of each station using a FastPrep DNA extraction kit for soil (MP Biomedicals, Santa Ana, CA, USA) and a FastPrep-24 cell disrupter (MP Biomedicals, Solon, OH, USA) according to the manufacturer’s instructions. The sediments were pretreated as previously described in order to remove humic acids and other interfering substances [33]. Sediments were then centrifuged at 4 °C for 10 min at 3000 rpm, and the supernatant was discarded. The dried sediments were used for downstream DNA extraction. Three separate extractions were conducted for each sample and the produced DNA was mixed. A NanoDrop 2000 (Thermo Scientific, Wilmington, DE, USA) spectrophotometer was used to measure the quantity and quality of the extracted DNA.
nirK sequences were amplified from the extracted DNA and the PCR reactions were incubated under the conditions shown in Table 1. However, for some samples, the concentration of MgCl2, template DNA, and primers were modified to optimize yields of the target product. Amplification products with predicted length were electrophoresed on a 1.0% TAE gel and then cloned into a pUCm-T vector (Promega) using the TOPO TA cloning kit (Invitrogen) according to the manufacturer’s instructions. Five libraries were constructed corresponding to surface sediments of each studied site. Approximately 50 positive clones were randomly taken from each library, and the correct insertions were checked by PCR amplification with primers FlaCu and R3Cu (Table 1) before finally sequencing.
Quantification of Denitrifying Microbial Groups
The abundance of denitrifying microbes was assessed by qPCR with different primers and thermal cycle parameters, as shown in Table 1. DNA from sediments at eight depths (0–1, 1–2, 2–3, 3–5, 7–8, 12–13, 22–23, and 32–33 cm, referred to as −0, −1, −2, −3, −7, −12, −22, and −32, respectively) was used as template for qPCR analysis. Standard curves were generated with serial dilutions of plasmids containing target gene, which was cloned from an environmental sediment sample. The standard curves displayed linearity (R 2 ≥ 0.980) between threshold cycle (CT) and log-transformed gene copy numbers. Concentrations of linearized plasmid DNA enzymatically digested were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). All qPCR assays were carried out in triplicate with an ABI Prism 7500 sequence detection system (Applied Biosystems). In all qPCR experiments, fluorescence signals were collected at 72 °C, and negative controls lacking template DNA were subject to the same qPCR procedures to detect any possible contamination. Agarose gel electrophoresis and melting-curve analyses were routinely used to confirm the specificity of the qPCR. Melting curves were obtained at 60 to 95 °C, with a read every 1 °C and holding for 1 s between reads. The resultant qPCR data were analyzed with the second derivative maximum method using the ABI Prism 7500 SDS software (version 1.4; Applied Biosystems). The amplification efficiencies were 93.5, 90.6, and 95.1% for narG, nirK, and nosZ, respectively.
Phylogenetic and Statistical Analysis
The nirK sequences were edited initially to exclude primer and vector sequences using Editseq embedded in DNAStar (version 5.0). The data set was filtered to include only sequences with correct primer sequences. Sequences were then clipped at 474 bp and translated into amino acids (158 aa) and aligned using ClustalX [38] in MEGA5 [39]. Sequences having in-frame stop codon (s) were removed [40]. ClustalX was employed to generate sequence identity matrices for each pair of the aligned deduced amino acid sequences [41]. A phylip-formatted distance matrix of the translated amino acid sequences was constructed using Prodist in Phylip 3.69, which served as an input to DOTUR [42] for operational taxonomic units (OTU) clustering at a 3% dissimilarity threshold. The coverage indices of gene diversity (Shannon-Wiener [H] and Simpson [D]) and richness (bias-corrected Chao1, SChao1), and rarefaction curves of each nirK clone library were also calculated using DOTUR.
The nirK-harboring community comparison was determined with weighted UniFrac environmental clustering and principal coordinate analysis (PCoA). The online UniFrac program (http://bmf2.colorado.edu/unifrac/index.psp) takes molecular evolutionary distances of sequences and their environmental occurrences for community similarity analysis. Correlations between nirK-harboring communities and environmental factors were analyzed by canonical correspondence analysis (CCA) using the software Canoco 4.5 [43]. Pearson correlation analysis (significance level 0.05) of the diversity and abundance of target functional genes (nirK, nosZ, and narG) with environmental factors were performed with the statistics software MINITAB 16 [44, 45].
A NirK sequence database for phylogenetic analysis was constructed by combining the sequences obtained in this study, their top-hit sequences of strain recovered [46] from the GenBank as determined through BLASTp searches. The phylogenetic trees were constructed by a neighbor-joining method using MEGA5 with the bootstrap support of 1000 replicates and two models, i.e., Poisson model and p-distance, to verify the precision of the tree. There was no significant difference between the trees constructed by these two models.
Nucleic Acid Sequence Accession Numbers
GenBank submission numbers for the sequences from this study are KP750695–KP750809.
Results
Environmental Conditions
The TOC (%) content of each site ranged from 0.55 ± 0.06 (ECS03) to 0.90 ± 0.07 (SYS01), and gradually reduced with depth (Table S1). The TN (%) content was almost the same at each site, ranged from 0.07 to 0.15, and had no significant change with depth. The C/N ratios were slightly higher at SYS sites than ECS sites. Compared to other sites, the δ13C (‰) showed more depleted value at ECS01. For δ15N (‰), the highest value was 5.50 ± 0.25 at ECS03, while the lowest value was 2.84 ± 0.59 at ECS01. The concentration of NO3 − was slightly higher at SYS sites than ECS sites. The concentration of NO2 − was higher at surface sediment of SYS sites and lowest at ECS01 site. The concentration of NH4 + increased with depth, highest at ECS01 ranging from 132.65 to 651.02 μmol L−1, and lowest at ECS02 ranging from 7.44 to 55.06 μmol L−1. The concentration of SiO3 2− ranged from 232.3 ± 29.2 (SYS01) to 399.0 ± 36.8 μmol L−1 (ECS02) and had a larger varied range with depth at ECS01 than other sites. In contrast to NH4 + and SiO3 2−, the concentrations of PO4 3− was lowest at ECS01. The salinity of bottom water at ECS01, ECS02, ECS03, SYS01, and SYS02 was 33.4, 34.4, 32.3, 30.3, and 31.1‰, respectively.
Diversity and Distribution of the NirK Dominant OTUs
Five nirK clone libraries were constructed for the surface sediment samples of ECMS, and 115 clones were identified to contain a valid nirK gene fragment, resulting in 49 unique protein sequences and 22 OTUs (Table 2). The values of library coverage (C) ranged from 57 to 70%, and the microbial diversity index was variable among sites. The diversity of nirK-harboring microbiota was highest at SYS01 and ECS02, and lowest at SYS02.
Among all the OTUs, six OTUs (OTU 4, 7, 11, 12, 8, 14) occurred predominantly in the mud deposit zones of ECMS, accounting for 27.0, 20.0, 8.7, 7.8, 7.0, and 6.1% of total sequences, respectively. These dominant OTUs had obvious regional distribution characteristics (Fig. 2). SYS01 was only dominated with OTU 7. SYS02 and ECS02 had a similar structure that occupied mostly by OTU 11 and OTU 12. ECS01 was dominated with OTU 7 and OTU 8. ECS03 was dominated with OTU 7 and OTU 14. OTU8 was mainly observed at ECS01. Although SYS sites were spatially adjacent and influenced by the same currents, there was a huge difference in their nirK-harboring microbial community composition.

The proportion of each dominant OTU in each station
Phylogeny of the NirK Sequences
The 22 obtained distinct NirK OTUs shared 40–96.3% identity with each other, whereas the amino acid sequences had 66 to 99% identity to the closest-match NirK sequences except for the primer regions by BLAST analysis in GenBank. The NirK sequences in ECMS sediments were more variable when compared with other environments, such as marine water column [23, 24], pelagic sediments [25], meadow and forest soils [47, 48], grassland soil [49], and cultivated soils [11, 50]. A part of dominant OTU amino acid sequences had low identities (<95%) with NirK sequences available in GenBank, such as OTU 4, 12, and 14, which were 87, 90, and 88% identical to the closest-match NirK sequences.
A neighbor-joining (NJ) phylogenetic tree based on NirK sequences was constructed with 22 NirK OTUs from all sampling sites (Fig. 3). Four clusters were detected from the phylogenetic tree. Most of the clones were located in Cluster I and IV that accounted for 37 and 49% of total sequences, respectively. Cluster I showed identity of >97% with their closest-match sequences from GenBank and were most closely related to Silicibacter sp. Cluster IV was related to Pseudomonas sp. with identity of <58% (Fig. S1). The closest-match sequences (identity ≥97%) recovered from GenBank were discovered in the San Francisco Bay estuary and Yellow River estuary [51, 52]. Most of the OTUs were <80% identical to proteins of cultivated strains, leading to difficulty for determining the lineage of OTUs.

Phylogenetic tree constructed by NirK sequences obtained from ECMS sediments. The tree was constructed by a neighbor-joining method with the p-distance substitution model in MEGA5 based on deduced amino acid sequences. The tree branch distances represent the amino acid substitution rate, and the scale bar represents the expected number of changes per homologous position. Bootstrap values (no less than 40%) of 1000 resamplings are shown at each node. The Ani A/Msp78 Moraxella catarrhalis RH4 sequence was used as an outgroup
Spatial Distribution of the nirK-Harboring Microbial Assemblages and Relationship with Environmental Factors
The heterogeneous distribution of nirK-harboring microbial communities was confirmed via Fast UniFrac PCoA. The results demonstrated that there were substantial differences among the nirK-harboring microbial assemblages in ECMS sediments, and the first two principal coordinates (P1 and P2) explained 89.9% of the total community variability. The nirK sequences of sampling sites from different mud zones were clearly separated from each other except that SYS01 and ECS01 were similar, although statistical testing is not feasible due to the lack of enough samples. Environmental characteristics might have a strong influence on the composition and distribution of the nirK-harboring microbial community (Fig. 4).

Ordination diagram of the Fast UniFrac weighted and normalized PCoA analysis of five mud deposits sites in ECMS. The analysis was conducted with deduced NirK amino acid sequence data. Shown is the plot of the first two principal coordinate axes (P1 and P2) for PCoA and the distributions of nirK-harboring microbial assemblages (designated with the sampling station names) in response to these axes
All OTUs were used in the CCA analysis to determine the influence of environmental factors on the nirK-harboring microbiota community composition. The first two CCA dimensions (CCA1 and CCA2) explained 71.4% of the total variance in the nirK-harboring microbial community composition (Fig. 5a). CCA analysis revealed that the community composition of nirK-harboring microbiota was related to multiple environmental factors. SYS02 and ECS02 sites were separated from other sites mainly by the concentration of TN, and NH4 + appeared to influence the nirK assemblages of site ECS01 (Fig. 5a). The six dominant OTUs were used for another CCA analysis to evaluate the relationship between environmental factors and dominant OTUs. The first and second dimensions explained 61.3 and 31.3% of the total variance, respectively. The results obtained according to CCA1 indicated that OTU 11, 12 positively correlated with TN, TOC, PO4 3−, NO2 −, and SiO3 2−. Opposite results were obtained for OTU 7, 14. The results explained by CCA2 indicated that OTU 8 positively correlated with NH4 +, C/N and negatively with δ15N, δ13C. Opposite results were obtained for OTU 14 (Fig. 5b).

CCA ordination plots for the first two principal dimensions showing environmentally related distributions. a Relationship between environmental parameters and nirK-harboring assemblages of the five mud deposit sites in ECMS; b relationship between environmental parameters and the six dominant OTUs across all sites
Abundance of Denitrifying Microorganisms and Variation Tendency of Abundance Ratios
Heterogeneous distributions of denitrifier abundance in different sediment layers of ECMS were exhibited by qPCR results (Fig. 6). Generally, the relative abundance of nirK in all sites was the highest (P < 0.01), followed by that of narG and nosZ. The abundance of denitrifiers was normally highest at the surface sediment and decreased with depth. According to the difference in environmental features of each site, different variation trends along with depth in each site were observed. Different from the other four sites, ECS03 had a gentle variation trend along with depth. ECS01 had the highest abundance of denitrifiers (P < 0.05), generally two to five times higher than other sites. Oppositely, SYS02 had the lowest denitrifier abundance (P < 0.05). The abundance of denitrifiers in SYS02 decreased more rapidly from 0 to 2 cm than the other sites; especially, the nirK gene decreased more than one order of magnitude. Compared to all the denitrifier quantities in the deep layer, SYS01 had a more obvious downtrend from 12 to 32 cm.

Quantitative analysis of functional genes (nirK, narG, nosZ) in mud deposits of ECMS by qPCR method. Different colors and nodes for lines represent different stations
The abundance ratios of different functional genes involved in each step of denitrifying process are shown in Fig. 7. The abundance ratios of qnarG/qnosZ (ARGZ) fluctuated in a range of 1 to 5 at different sites and depths, which might imply a high potential of N2O/NO production instead of N2. The abundance of nitrite reductase qnirK was significantly higher than that of nitrate reductase (qnarG) (P < 0.01, Fig. 7), indicating that nitrite had a higher transformation efficiency than nitrate in the examined marine sediments.

The ratios of denitrifiers in the different sediment layers of ECMS. a SYS01; b SYS02; c ECS01; d ECS02; e ECS03
The Influence of Environmental Factors on Denitrifier Abundance and Diversity
The relationship between environmental factors and denitrifier abundance demonstrated that the response of denitrifiers to factors was not invariable in different mud deposit zones as proxies for various environmental conditions. The abundance of all the functional genes, including qnarG, qnirK, and qnosZ, was significantly positively correlated with TOC% and negatively with NH4 + and δ13C at site SYS01, while the quantities of nitrite reducer (qnirK) were positively correlated with NO2 − and SiO3 2− (P < 0.05) (Table 3). At SYS02, the abundance of all the targeted genes had significantly positive correlations with NO2 − (except for qnarG) and TN%, and negative correlations with NH4 + and SiO3 2−. Moreover, the functional gene abundance at ECS01 was significantly positively correlated with TOC% and negatively with NH4 +, SiO3 2−, and PO4 3−. However, we found no significant correlations between almost all of the environment factors with the quantities of denitrifier at ECS02. The data of environmental factors were not intact at ECS03, but an interesting phenomenon could be found that qnosZ was negatively correlated with TOC% and TN%. Additionally, environmental factors had no significant impacts on diversity of nirK-harboring microorganisms (P > 0.05), and only NH4 + had a significant impact (P < 0.05) on abundance of nirK-harboring microorganisms (Table S2).
ARGZ was significantly correlated with NH4 +, SiO3 2−, PO4 3−, and TOC% at ECS01, with TN% and C/N at SYS02, and with TN% at ECS03 (Table 4). Unexpectedly, we found no regular relationships between the abundance ratios of each functional gene and environmental factors. The results demonstrated that in different situations, the response of microbiota composition and diversity on environmental factors was different.
Discussion
Diversity and Distribution of the nirK Gene Sequences
The nirK-harboring microbiota were dominated by a few OTUs, and they were found to be widely distributed in different sites with each site harboring their unique phylotypes. This was confirmed by the results of PCoA analysis in which sites from different mud areas were clearly separated. This could have resulted from the low dissimilarity threshold (3%) used for clustering the NirK amino acid sequences which would inflate the calculation of diversity indices. However, a fine-scale clustering threshold would enable to gain more accurate taxonomical and functional distributions. Similar to our results, a previous study reported the site-specific differences in denitrifier community compositions from soils [53]. Every single site from the same sea area could represent a potential ecological niche for microbes, where the microenvironmental conditions that constrained and enriched different denitrifier communities under primary succession would be a possible explanation for this result. Furthermore, the OTUs from the same sampling site also had different responses to environmental factors even if they were phylogenetically closer to each other. For example, OTU 11 had a positive correlation with SiO3 2−, TN, PO4 3−, TOC, and NO2 −, while OTU 14 had an opposite result (Fig. 5). The results suggested that marine microbes from the same lineage could have different ecological functions.
A phylogenetic tree was constructed by combination of the 22 NirK OTUs retrieved in our study and closest-match cultivated strain sequences from the NCBI. All the 22 OTUs were distinct from their known closest reference strain sequences in GenBank with identities far less than 97%, indicating that many denitrifiers ubiquitous in marine sediments are still unknown, which need further exploration. From the tree, the sequences could be classified into four clusters (I to IV), and the dominant OTUs were primarily grouped into Cluster I and IV (Fig. 3). Cluster IV was of interest because sequences of this clade were clustered with a high bootstrap value (Fig. 3). Although sequences of Cluster IV showed an identity value greater than 97% with their closest-match sequences from GenBank, they had very low identity value with cultivated reference strains (most related to Pseudomonas sp. PI1 (WP_052267715) with identities less than 58%), suggesting that an abundant clade of denitrifiers is prevalent in the surface sediment of the mud areas of ECMS, but their physiological features are yet to be characterized.
The results of diversity index analysis demonstrated that the richness and diversity of denitrifiers in mud deposit zones of ECMS were lower than the ones in the Jiaozhou Bay [54] and Yangtze Estuary [55], and higher than the ones in the surface sediment of the South China Sea [25, 56], although the sequencing depth was not enough to cover the local denitrifying community. Sedimentary organic matter might explain this discrepancy since sites closer to land usually receive more inputs from land and therefore have more organic matter deposition, although comparison of denitrifier communities in these studies is not feasible due to the lack of common environmental factors.
Denitrifier Abundance in Different Layers and Relationship with Environmental Factors
The abundance of nirK (2.47 × 106 to 6.49 × 108 copies g−1, dry weight) was significantly higher than that in deep-sea subsurface sediments of the South China Sea (1.30 × 103 to 1.74 × 105 copies g−1, dry weight) [25] or sediments of hypernutrified estuarine tidal flats (2.7 × 103 to 1.6 × 107 copies g−1, wet weight) [57]. The abundance of denitrifiers was significantly higher than that of anammox bacteria (unpublished data) and one to two orders of magnitude higher than that of ammonium oxidation organisms measured with amoA in our previous study targeting the same sites [19], demonstrating that denitrification is a more prevalent process compared with anammox and ammonium oxidation in the mud deposit zones in which the high concentration of organic matter might have facilitated the enrichment of denitrifiers [36]. Furthermore, the changing trends of abundance of the three functional genes were different from that of amoA in each site and different layers [19], suggesting a decoupling of ammonia oxidation and denitrification in the sediments. However, there were not any consistent correlations between the abundance of denitrifiers and the environmental factors including nitrate and nitrite across all sites, indicating that each mud zone could have significantly distinct environmental characteristics.
To the best of our knowledge, this was the first study to examine the denitrifier abundance in different sediment layers of ECMS. The abundance of denitrifiers was the highest at the surface sediment and decreased with depth in all studied sites. The abundance of denitrifiers in SYS02 sediments except for the surface (0 cm) was much lower than other sites (Fig. 6). The high concentration of methane existed at SYS02 site (unpublished data) might benefit the growth of Methanotroph that competed with denitrifiers and inhibited the multiplication of denitrifiers. ECS01 site located just outside of the Yangtze Estuary was largely affected by the river runoff and had the highest abundance of denitrifiers. The denitrifiers’ abundance in ECS01 had a significant correlation with δ15N, illustrating that the source of organic nitrogen may have significant impacts on the denitrifying process.
In addition, three functional genes were quantified in the present study to gain a better understanding of the abundance profiles of denitrifiers [15, 58]. Generally, the marine mud deposit zones (SYS01, SYS02, ECS01, ECS02, ECS03) had a higher abundance of nitrite reductase gene than that of nitrate and nitrous oxide reductase genes, which might suggest that nitrite reduction occurs more frequently than nitrate and nitrous oxide reduction. This result indicated that nitrite might be reduced independently of denitrification in marine sediments. The variation of ARGZ (ranging from 1 to 5) was observed across all samples. ARGZ was the highest at site ECS01, and ARGZ at the sites near continent (ECS01, ECS02) was apparently higher than that in the open sea sites (SYS01, SYS02, ECS03). Hence, consistent with the results mentioned above, the distance of sites to the land, as a proxy of anthropogenic activities and flow out of freshwater, was an important factor to drive the composition and distribution of the denitrifier community in mud deposit zones of ECMS systems. The high ARGZ value demonstrated that nitrate might have a higher potential to be transformed into greenhouse gases [59]. However, N2O could be reduced to N2 by a non-canonical denitrification pathway via distinct “atypical” NosZ protein [60].
It was interesting to find that the abundance of each denitrifying functional gene varied greatly in a single deposit zone, indicating that many microbes did not harbor a complete set of genes relevant to denitrification. In addition, a great diversity of denitrifiers was found in estuarine and coastal sediments [61]. The higher diversity of microbes participating in the denitrification process together with the unbalance of different denitrification reactions gave a hint that the denitrifying processes were accomplished through the cooperation of different denitrifying microorganisms in the marine sediment.
In conclusion, the present study provided an outline of denitrifying microbiota in mud deposit zones of ECMS. Each mud area had its own denitrifier assemblages, and these site-specific differences in denitrifier communities might be attributed to anthropogenic activities and freshwater runoff, which have great effects on the mud deposit zones of ECMS systems. In addition, a few phylogenetic closely related OTUs were found to have different responses to environmental factors, implicating different roles they played in the coastal sedimentary ecosystems. A significantly higher abundance of nirK gene was observed than that of narG and nosZ genes, as evidenced by their abundance ratios. The inconsistent variations of different functional genes in the sites examined suggested that multiple genes should be considered to gain a more comprehensive insight into the distribution of denitrifiers in future studies. Finally, the three denitrifying functional genes displayed different changing trends from the amoA gene in each site and different layers, suggesting a decoupling of ammonia oxidation and denitrification in the sediments.
References
Gruber N (2008) The marine nitrogen cycle: overview and challenges. In: Douglas GC (ed) Nitrogen in the marine environment, 2nd edn. Academic Press, pp 1–50
Philippot L, Spor A, Hénault C, Bru D, Bizouard F, Jones CM, Sarr A, Maron P-A (2013) Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7:1609–1619. doi:10.1038/ismej.2013.34
Bowen JL, Weisman D, Yasuda M, Jayakumar A, Morrison HG, Ward BB (2015) Marine oxygen-deficient zones harbor depauperate denitrifying communities compared to novel genetic diversity in coastal sediments. Microb Ecol 70:311–321. doi:10.1007/s00248-015-0582-y
Yao P, Zhao B, Bianchi TS, Guo Z, Zhao M, Li D, Pan H, Wang J, Zhang T, Yu Z (2014) Remineralization of sedimentary organic carbon in mud deposits of the Changjiang Estuary and adjacent shelf: implications for carbon preservation and authigenic mineral formation. Cont Shelf Res 91:1–11. doi:10.1016/j.csr.2014.08.010
Knowles R (1982) Denitrification. Microbiol Rev 46:43–70
Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616
Bru D, Sarr A, Philippot L (2007) Relative abundances of proteobacterial membrane-bound and periplasmic nitrate reductases in selected environments. Appl Environ Microbiol 73:5971–5974. doi:10.1128/AEM.00643-07
Braker G, Zhou J, Wu L, Devol AH, Tiedje JM (2000) Nitrite reductase genes (nirK and nirS) as functional markers to investigate diversity of denitrifying bacteria in Pacific Northwest marine sediment communities. Appl Environ Microbiol 66:2096–2104. doi:10.1128/AEM.66.5.2096-2104.2000
Horn MA, Drake HL, Schramm A (2006) Nitrous oxide reductase genes (nosZ) of denitrifying microbial populations in soil and the earthworm gut are phylogenetically similar. Appl Environ Microbiol 72:1019–1026. doi:10.1128/AEM.72.2.1019-1026.2006
Huang S, Chen C, Yang X, Wu Q, Zhang R (2011) Distribution of typical denitrifying functional genes and diversity of the nirS-encoding bacterial community related to environmental characteristics of river sediments. Biogeosciences 8:3041–3051. doi:10.5194/bg-8-3041-2011
Dandie CE, Wertz S, Leclair CL, Goyer C, Burton DL, Patten CL, Zebarth BJ, Trevors JT (2011) Abundance, diversity and functional gene expression of denitrifier communities in adjacent riparian and agricultural zones. FEMS Microbiol Ecol 77:69–82. doi:10.1111/j.1574-6941.2011.01084.x
Chon K, Cho J (2015) Abundance and expression of denitrifying genes (narG, nirS, norB, and nosZ) in sediments of wastewater stabilizing constructed wetlands. Environ Eng Res 20:51–57. doi:10.4491/eer.2014.069
Bárta J, Melichová T, Vaněk D, Picek T, Šantrůčková H (2010) Effect of pH and dissolved organic matter on the abundance of nirK and nirS denitrifiers in spruce forest soil. Biogeochemistry 101:123–132. doi:10.1007/s10533-010-9430-9
Crutzen PJ (1979) The role of NO and NO2 in the chemistry of the troposphere and stratosphere. Annu Rev Earth Planet Sci 7:443–472
Henry S, Bru D, Stres B, Hallet S, Philippot L (2006) Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl Environ Microbiol 72:5181–5189. doi:10.1128/AEM.00231-06
Smith CJ, Nedwell DB, Dong LF, Osborn AM (2007) Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Appl Environ Microbiol 73:3612–3622. doi:10.1128/AEM.02894-06
Liu J, Xu K, Aea L, Milliman J, Velozzi D, Xiao S, Yang Z (2007) Flux and fate of Yangtze River sediment delivered to the East China Sea. Geomorphology 85:208–224. doi:10.1016/j.geomorph.2006.03.023
Hu L, Shi X, Guo Z, Wang H, Yang Z (2013) Sources, dispersal and preservation of sedimentary organic matter in the Yellow Sea: the importance of depositional hydrodynamic forcing. Mar Geol 335:52–63. doi:10.1016/j.margeo.2012.10.008
Yu S, Yao P, Liu J, Zhao B, Zhang G, Zhao M, Yu Z, Zhang X-H (2016) Diversity, abundance, and niche differentiation of ammonia-oxidizing prokaryotes in mud deposits of the eastern China marginal seas. Front Microbiol 7:137. doi:10.3389/fmicb.2016.00137
Magalhães C, Bano N, Wiebe W, Bordalo A, Hollibaugh J (2008) Dynamics of nitrous oxide reductase genes (nosZ) in intertidal rocky biofilms and sediments of the Douro River Estuary (Portugal), and their relation to N-biogeochemistry. Microb Ecol 55:259–269. doi:10.1007/s00248-007-9273-7
Chon K, Chang J-S, Lee E, Lee J, Ryu J, Cho J (2011) Abundance of denitrifying genes coding for nitrate (narG), nitrite (nirS), and nitrous oxide (nosZ) reductases in estuarine versus wastewater effluent-fed constructed wetlands. Ecol Eng 37:64–69. doi:10.1016/j.ecoleng.2009.04.005
Wu L, Osmond DL, Graves AK, Burchell MR, Duckworth OW (2012) Relationships between nitrogen transformation rates and gene abundance in a riparian buffer soil. Environ Manag 50:861–874. doi:10.1007/s00267-012-9929-z
Jayakumar DA, Francis CA, Naqvi SWA, Ward BB (2004) Diversity of nitrite reductase genes (nirS) in the denitrifying water column of the coastal Arabian Sea. Aquat Microb Ecol 34:69–78. doi:10.3354/ame034069
Ward B, Devol A, Rich J, Chang B, Bulow S, Naik H, Pratihary A, Jayakumar A (2009) Denitrification as the dominant nitrogen loss process in the Arabian Sea. Nature 461:78–81. doi:10.1038/nature08276
Li M, Hong Y, Cao H, Klotz MG, Gu JD (2013) Diversity, abundance, and distribution of NO-forming nitrite reductase-encoding genes in deep-sea subsurface sediments of the South China Sea. Geobiology 11:170–179. doi:10.1111/gbi.12020
Vilar-Sanz A, Puig S, García-Lledó A, Trias R, Balaguer MD, Colprim J, Bañeras L (2013) Denitrifying bacterial communities affect current production and nitrous oxide accumulation in a microbial fuel cell. PLoS ONE 8:e63460. doi:10.1371/journal.pone.0063460
Hu L, Shi X, Yu Z, Lin T, Wang H, Ma D, Guo Z, Yang Z (2012) Distribution of sedimentary organic matter in estuarine–inner shelf regions of the East China Sea: implications for hydrodynamic forces and anthropogenic impact. Mar Chem 142:29–40. doi:10.1016/j.marchem.2012.08.004
Chen Z, Saito Y, Kanai Y, Wei T, Li L, Yao H, Wang Z (2004) Low concentration of heavy metals in the Yangtze estuarine sediments, China: a diluting setting. Estuar Coast Shelf Sci 60:91–100. doi:10.1016/j.ecss.2003.11.021
Huh C-A, Su C-C (1999) Sedimentation dynamics in the East China Sea elucidated from 210Pb, 137Cs and 239,240 Pu. Mar Geol 160:183–196. doi:10.1016/S0025-3227(99)00020-1
Liu J, Zhu R, Li G (2003) Rock magnetic properties of the fine-grained sediment on the outer shelf of the East China Sea: implication for provenance. Mar Geol 193:195–206. doi:10.1016/S0025-3227(02)00660-6
Yang SY, Jung HS, Lim DI, Li CX (2003) A review on the provenance discrimination of sediments in the Yellow Sea. Earth-Sci Rev 63:93–120. doi:10.1016/S0012-8252(03)00033-3
Lim D, Choi J, Jung H, Rho K, Ahn K (2007) Recent sediment accumulation and origin of shelf mud deposits in the Yellow and East China Seas. Prog Oceanogr 73:145–159. doi:10.1016/j.pocean.2007.02.004
Throbäck IN, Enwall K, Jarvis Å, Hallin S (2004) Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49:401–417. doi:10.1016/j.femsec.2004.04.011
Hallin S, Lindgren PE (1999) PCR detection of genes encoding nitrite reductase in denitrifying bacteria. Appl Environ Microbiol 65:1652–1657
Hallin S, Jones CM, Schloter M, Philippot L (2009) Relationship between N-cycling communities and ecosystem functioning in a 50-year-old fertilization experiment. ISME J 3:597–605. doi:10.1038/ismej.2008.128
Hill AR, Cardaci M (2004) Denitrification and organic carbon availability in riparian wetland soils and subsurface sediments. Soil Sci Soc Am J 68:320–325. doi:10.2136/sssaj2004.3200
Liu J, Hou H, Sheng R, Chen Z, Zhu Y, Qin H, Wei W (2012) Denitrifying communities differentially respond to flooding drying cycles in paddy soils. Appl Soil Ecol 62:155–162. doi:10.1016/j.apsoil.2012.06.010
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Farnelid H, Bentzon-Tilia M, Andersson AF, Bertilsson S, Jost G, Labrenz M, Jürgens K, Riemann L (2013) Active nitrogen-fixing heterotrophic bacteria at and below the chemocline of the central Baltic Sea. ISME J 7:1413–1423. doi:10.1038/ismej.2013.26
Larkin MA, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi:10.1093/bioinformatics/btm404
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506. doi:10.1128/AEM.71.3.1501-1506.2005
Ter Braak CJ, Smilauer P (2002) CANOCO reference manual and CanoDraw for Windows user’s guide: software for canonical community ordination (version 4.5). www. canoco. com
Dang H, Chen R, Wang L, Guo L, Chen P, Tang Z, Tian F, Li S, Klotz MG (2010) Environmental factors shape sediment anammox bacterial communities in hypernutrified Jiaozhou Bay, China. Appl Environ Microbiol 76:7036–7047. doi:10.1128/AEM.01264-10
Dang H, Luan X-W, Chen R, Zhang X, Guo L, Klotz MG (2010) Diversity, abundance and distribution of amoA-encoding archaea in deep-sea methane seep sediments of the Okhotsk Sea. FEMS Microbiol Ecol 72:370–385. doi:10.1111/j.1574-6941.2010.00870.x
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585. doi:10.1128/AEM.01996-06
Rösch C, Mergel A, Bothe H (2002) Biodiversity of denitrifying and dinitrogen-fixing bacteria in an acid forest soil. Appl Environ Microbiol 68:3818–3829. doi:10.1128/AEM.68.8.3818-3829.2002
Guo G-X, Deng H, Qiao M, Yao H-Y, Zhu Y-G (2013) Effect of long-term wastewater irrigation on potential denitrification and denitrifying communities in soils at the watershed scale. Environ Sci Technol 47:3105–3113. doi:10.1021/es304714a
Hartmann AA, Barnard RL, Marhan S, Niklaus PA (2013) Effects of drought and N-fertilization on N cycling in two grassland soils. Oecologia 171:705–717. doi:10.1007/s00442-012-2578-3
Yoshida M, Ishii S, Otsuka S, Senoo K (2010) nirK-harboring denitrifiers are more responsive to denitrification-inducing conditions in rice paddy soil than nirS-harboring bacteria. Microbes Environ 25:45–48. doi:10.1264/jsme2.ME09160
Mosier AC, Francis CA (2010) Denitrifier abundance and activity across the San Francisco Bay estuary. Environ Microbiol Rep 2:667–676. doi:10.1111/j.1758-2229.2010.00156.x
Li J, Wei G, Wang N, Gao Z (2014) Diversity and distribution of nirK-harboring denitrifying bacteria in the water column in the Yellow River Estuary. Microbes Environ 29:107–110. doi:10.1264/jsme2.ME13111
Rich J, Heichen R, Bottomley P, Cromack K, Myrold D (2003) Community composition and functioning of denitrifying bacteria from adjacent meadow and forest soils. Appl Environ Microbiol 69:5974–5982. doi:10.1128/AEM.69.10.5974-5982.2003
Dang H, Wang C, Li J, Li T, Tian F, Jin W, Ding Y, Zhang Z (2009) Diversity and distribution of sediment nirS-encoding bacterial assemblages in response to environmental gradients in the eutrophied Jiaozhou Bay, China. Microb Ecol 58:161–169. doi:10.1007/s00248-008-9469-5
Zheng Y, Hou L, Liu M, Gao J, Yin G, Li X, Deng F, Lin X, Jiang X, Chen F (2015) Diversity, abundance, and distribution of nirS-harboring denitrifiers in intertidal sediments of the Yangtze Estuary. Microb Ecol 70:30–40. doi:10.1007/s00248-015-0567-x
Katsuyama C, Kondo N, Suwa Y, Yamagishi T, Itoh M, Ohte N, Kimura H, Nagaosa K, Kato K (2008) Denitrification activity and relevant bacteria revealed by nitrite reductase gene fragments in soil of temperate mixed forest. Microbes Environ 23:337–345. doi:10.1264/jsme2.ME08541
Zhang X, Agogué H, Dupuy C et al (2014) Relative abundance of ammonia oxidizers, denitrifiers, and anammox bacteria in sediments of hyper-nutrified estuarine tidal flats and in relation to environmental conditions. [J] CLEAN–Soil, Air, Water 42(6):815–823. doi:10.1002/clen.201300013
Chen Z, Liu J, Wu M, Xie X, Wu J, Wei W (2012) Differentiated response of denitrifying communities to fertilization regime in paddy soil. Microb Ecol 63:446–459. doi:10.1007/s00248-011-9909-5
Voss M, Bange HW, Dippner JW, Middelburg JJ, Montoya JP, Ward B (2013) The marine nitrogen cycle: recent discoveries, uncertainties and the potential relevance of climate change. Philos Trans R Soc B 368. doi: 10.1098/rstb.2013.0121
Orellana LH, Rodriguez-R LM, Higgins S, Chee-Sanford JC, Sanford RA, Ritalahti KM, Löffler FE, Konstantinidis KT (2014) Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. MBio 5:e01193–14. doi:10.1128/mBio.01193-14
Jetten MS (2008) The microbial nitrogen cycle. Environ Microbiol 10:2903–2909. doi:10.1111/j.1462-2920.2008.01786.x
Acknowledgements
We are grateful to the captain and crew of the R/V Dong Fang Hong 2 for their assistance during the cruise. We also appreciate all colleagues who contributed to this study. This work was supported by the National Natural Science Foundation of China through grants 41521064, 41476112, and 41506154.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Minghong Gao and Jiwen Liu contributed equally to this work.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 327 kb)
Rights and permissions
About this article

Cite this article
Gao, M., Liu, J., Qiao, Y. et al. Diversity and Abundance of the Denitrifying Microbiota in the Sediment of Eastern China Marginal Seas and the Impact of Environmental Factors. Microb Ecol 73, 602–615 (2017). https://doi.org/10.1007/s00248-016-0906-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-016-0906-6