
Abstract
Denitrification plays a critical role in nitrogen removal in estuarine and coastal ecosystems. In this study, the community composition, diversity, abundance, and distribution of cytochrome cd1-type nitrite reductase gene (nirS)-harboring denitrifiers in intertidal sediments of the Yangtze Estuary were analyzed using polymerase chain reaction (PCR)-based clone libraries and quantitative PCR techniques. Clone library analysis showed that the nirS-encoding bacterial biodiversity was significantly higher at the lower salinity sites than at the higher salinity sites. However, there was no significant seasonal difference in the nirS gene diversity between summer and winter. Phylogenetic analysis revealed that the nirS-harboring denitrifier communities at the study area had distinctive spatial heterogeneity along the estuary. At the lower salinity sites, the nirS-harboring bacterial community was co-dominated by clusters III and VII; while at the higher salinity sites, it was dominated by cluster I. Canonical correspondence analysis indicated that the community compositions of nirS-type denitrifiers were significantly correlated with salinity, ammonium, and nitrate. Quantitative PCR results showed that the nirS gene abundance was in the range of 1.01 × 106 to 9.00 × 107 copies per gram dry sediment, without significant seasonal variation. Among all the environmental factors, the nirS gene abundance was only significantly related to the change of salinity. These results can extend our current knowledge about the composition and dynamics of denitrification microbial community in the estuarine ecosystem.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Global nitrogen (N) overload has been identified as a main emerging environmental issue in this century, due mainly to the excessive anthropogenic N input [1, 2]. Much of the anthropogenic N is transported into estuarine and coastal regions, which has already exerted a serious threat to the environmental quality of estuarine and coastal ecosystems [2]. However, microorganism-mediated denitrification is capable of removing significant quantities of the excessive N load (>50 % of which is nitrate) from these N-enriched environments, providing a sink for N, and thereby, playing an important role in decreasing the degree of eutrophication [3–6]. Denitrification is the dissimilatory reduction of nitrate and nitrite to gaseous products (NO, N2O, and N2) under suboxic conditions. It is a major biological loss term for fixed N from terrestrial and aquatic ecosystems to the atmosphere, especially in estuaries, where it was estimated to contribute up to 93.4 % to the total nitrogen loss while the anaerobic oxidation of ammonium to nitrogen gas (anammox) was much less quantitatively significant [7, 8]. Considering the importance of denitrification for nitrogen removal, it is critical to understand the community dynamics and distribution of the underlying denitrifiers in estuarine ecosystems.
Denitrification proceeds by a diverse assemblage of microorganisms, encoding different types of metabolic enzymes, such as nitrate reductases, nitrite reductases (Nir), nitric oxide reductases, and nitrous oxide reductases (Nos) [9]. Nitrite reduction to nitric oxide, catalyzed by either cytochrome cd-1 NirS nitrite reductase or copper-containing NirK enzymes, is the rate-limiting step in denitrification [9]. Besides, Nir distinguishes the true denitrifiers (gas producing) from nitrate-respiring microbes (including those performing dissimilatory nitrate or nitrite reduction to ammonium) [9], and thus, cytochrome cd1-type nitrite reductase (nirS) and nirK (functionally equivalent but structurally different) genes have been most frequently targeted for molecular diversity studies of denitrifiers [6]. However, nirS gene is much more commonly used than nirK gene in estuarine systems as it is reported that cytochrome cd-1 NirS nitrite reductase is far more abundant than NirK in estuarine environments [6, 9–11].
In the present study, we selected the Yangtze Estuary as our study area to explore the diversity, abundance, and distribution of denitrifiers in estuarine ecosystems based on nirS gene. The Yangtze Estuary located in the subtropical monsoon climate zone is China’s largest estuary, covering an area of about 8,500 km2. The Yangtze River delivers more than 7.5 × 1010 moles of N nutrients per year to the East China Sea through the estuarine regions [12]. Especially in recent decades, the Yangtze Estuary has been receiving an increasing load of anthropogenic nitrogen from fish farming, agricultural activities, and both industrial and domestic wastewater discharge, which has resulted in severely eutrophic status in the estuarine and adjacent coastal area [13, 14]. Therefore, the microbial nitrogen removal is of major concern in the Yangtze Estuary. To date, however, the molecular dynamics and distribution of nitrite reductase genes in intertidal sediments along the Yangtze Estuary still remains unexplored. Previously, we have examined the anammox bacterial communities [8] and ammonia-oxidizing prokaryotic communities [15] in these sediments of the Yangtze Estuary, showing distinctive spatial heterogeneities along the estuary which were correlated significantly with salinity. While a similar pattern might be expected for the distribution of nirS-harboring denitrifiers along the estuary as the latter were tightly linked with anammox bacterial and ammonia-oxidizing communities, the diversity and abundance of denitrifiers might also be related to key environmental factors such as reactive nitrogen and organic carbon which most denitrifiers were dependent on [9].
The objectives of this study were (a) to investigate the diversity, community composition, and abundance of denitrifiers in the Yangtze Estuary based on the nirS gene, (b) to elucidate the distribution pattern of nirS-harboring denitrifiers in the estuarine ecosystem, and (c) to explore potential links of estuarine environmental variables with the dynamics of nirS-harboring denitrifiers.
Materials and Methods
Field Sampling
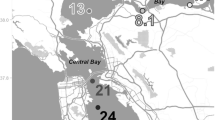
In this study, surface sediment samples were collected from seven representative sites from the intertidal flats along the Yangtze Estuary (Fig. 1), including Xupu (XP), Liuhekou (LHK), Wusongkou (WSK), Bailonggang (BLG), Daxingang (DXG), Yinyang (YY), and Luchao (LC). Field surveys were conducted in January (winter) and August (summer) 2011, respectively. At each site, triplicate surface sediments (0–5 cm) were collected with stainless steel tubes and shovels. The sediment samples were then stored in sterile plastic bags, sealed and transported to the laboratory on ice within 4 h. Upon return to the laboratory, triplicate surface sediments from each site were homogenized immediately under a nitrogen atmosphere as one composite sample. Subsequently, one part of the sample from each site was preserved at −80 °C for deoxyribonucleic acid (DNA) extraction and subsequent molecular analysis. The other part was stored at 4 °C for denitrification rate measurements and sediment physiochemical analyses.

The study area showing the sampling sites
The denitrification rates and environmental parameters in association with these sediment samples from the Yangtze Estuary were analyzed and have been shown in our previous publication (Supplementary Fig. S1 and Table S1) [8].
DNA Extraction, Gene Amplification, Cloning, and Phylogenetic Analysis
Total genomic DNA was extracted from duplicate composite sediment samples (∼1.0 g) using Ultraclean™ soil DNA Isolation Kits (MOBIO, USA) following the manufacturer’s instructions. The nirS gene fragments (∼840–890 bp) were amplified from pooled sediment DNA extracts using the polymerase chain reaction (PCR) primers nirS-1F (5′-CCTAYTGGCCGCCRCART-3′) and nirS-6R (5′-CGTTGAACTTRCCGGT-3′) [16]. PCR was performed in a total volume of 50 μL containing 1 μL of Taq DNA Polymerase (5 U μL−1, Sangon), 5 μL of 10 × PCR buffer (without MgCl2, Sangon, China), 4 μL of MgCl2 (25 mM, Sangon), 1 μL of dNTP (each 10 mM, Sangon), 1 μL of forward primer (10 μM, Sangon), 1 μL of reverse primer (10 μM, Sangon), and 1 μL of genomic DNA. PCR reactions were carried out using a thermal program of 95 °C for 5 min, 30 cycles of 95 °C for 30 s, 56 °C for 40 s, and 72 °C for 40 s, and a final extension cycle at 72 °C for 5 min.
Appropriately sized nirS gene fragments were confirmed and separated by electrophoresis in 1 % agarose gel and purified using Gel Advance-Gel Extraction system (Viogene, China). The purified products were cloned using the TOPO-TA cloning kit (Invitrogen, USA) in accordance with the manufacturer’s instructions. Clones were randomly selected for sequencing with an ABI Prism 3730 Genetic Analyzer (Applied BioSystems, USA) using Sanger method. The obtained nirS sequences were edited using DNAstar software package (DNASTAR, USA), and possible chimeras were checked using the CHECK CHIMERA program of the Ribosomal Database Project [17]. The unique nirS gene sequences obtained in this study are available in GenBank under accession numbers KM891747 to KM892364. The nirS gene sequences were analyzed initially using the BLASTn tool (http://www.ncbi.nlm.nih.gov/BLAST/) to aid the selection of the closest matches. All the sequences and their closest matches obtained from the NCBI were aligned by ClustalX program (version 2.1) [18]. The sequences with more than 95 % identity were grouped into one operational taxonomic unit (OTU) using Mothur (http://www.mothur.org/wiki/Main_Page) by the furthest neighbor approach [19]. Neighbor-joining phylogenetic tree was constructed using MEGA software (version 5.03) [20], and the relative confidence of the tree topologies was evaluated by performing 1,000 bootstrap replicates [21].
Real-Time Quantitative PCR
Plasmids carrying a nirS gene fragment were extracted from Escherichia coli hosts using a Plasmid Mini Preparation Kit (Tiangen, China) for standard curve construction. Plasmid DNA concentrations were measured using a Nanodrop-2000 Spectrophotometer (Thermo, USA). Triplicate samples and standard reactions were performed with an ABI 7500 Sequence Detection System (Applied Biosystems, Canada). The primer set composed of cd3aF (5′-GTSAACGTSAAGGARACSGG-3′) and R3cd (5′-GASTTCGGRTGSGTCTTGA-3′) was used to quantify the nirS gene (The size of the PCR products is 425 bp.) [22]. The quantitative PCR (qPCR) was performed in a total volume of 25 μL containing 12.5 μL of Maxima SYBR Green/Rox qPCR Master Mix (Fermentas, Lithuania), 1 μL of template DNA, and 1 μL of each primer (10 μM). The specificity of the qPCR amplification was determined by the melting curve and gel electrophoresis. The qPCR protocol was as follows: 2 min at 50 °C, 10 min at 95 °C, followed by 45 cycles of 95 °C for 30 s, 58 °C for 40 s, and 72 °C for 1 min. Negative controls containing no template DNA were subjected to detect and exclude any possible contamination. The nirS gene abundance was calculated based on the constructed standard curve, and then converted into copies per gram of sediment, assuming the DNA extraction efficiency was 100 %.
Statistical Analysis
The Mothur program was used to generate rarefaction curves for the observed OTUs and to determine the species richness Chao1 estimator and diversity indices (Shannon-Weiner and Simpson indices) [19]. The coverage of nirS gene clone libraries was calculated by dividing the observed number of OTUs by Chao1 estimator [23]. Correlations between nirS-encoding bacterial assemblages and environmental parameters were explored with the canonical correspondence analysis (CCA; the maximum gradient length was 4SD based on the detrended correspondence analysis, showing that the responses of OTUs to environmental variables were unimodal) using the software Canoco (version 4.5) [24]. Community classification of the sediment nirS-harboring denitrifiers was performed via principal coordinates analysis (PCoA) using the UniFrac program (http://bmf.colorado.edu/unifrac/index.psp) [25, 26]. Also, UniFrac was used to form the distance matrix of the constructed clone libraries, and all P values have been corrected for multiple comparisons by multiplying them by the number of comparisons that were made (the Bonferroni correction) [25, 26].
Results
Diversity of nirS Clone Libraries
The nirS gene fragments were successfully amplified from all the sampling sites of the Yangtze Estuary in both summer and winter (Supplementary Fig. S2). A total of 14 nitrite-reducing bacterial nirS gene clone libraries were generated, and overall 1,840 sequences were obtained for further analysis (Table 1). When 5 % divergence in nucleotides was used as the cut-off value, 15 to 42 OTUs were obtained within each individual clone library. Rarefaction analysis as well as the Shannon-Weiner and Simpson indices showed that the greatest nirS gene biodiversity was observed at the lower salinity site XP in summer, while the lowest value was occurred at the higher salinity site DXG in winter (Fig. 2 and Table 1). Generally, the diversity of nirS gene was significantly higher at the lower salinity sites than at the higher salinity sites in the intertidal sediments of the Yangtze Estuary (Student’s t test, P = 0.01). However, no obvious seasonal difference in the nirS gene diversity between summer and winter was observed (Student’s t test, P > 0.05) (Supplementary Table S2). The estimated coverage values of the clone libraries were between 88.2 and 96.6 %, indicating that the majority of the nirS sequence types were captured, and this was further confirmed by the gradually flattening rarefaction curves (Table 1 and Fig. 2).

Rarefaction analysis of nirS-harboring denitrifier communities in the marsh sediments of the Yangtze Estuary. OTUs are defined by <5 % divergence in nucleotide sequence. S and W before the hyphen character (−) represent summer and winter, respectively
Phylogenetic Analysis of nirS Sequences
In addition to comparing the relative richness and biodiversity of the nirS gene, we examined the phylogenetic relationships of these sequences. For the purposes of discussion, we have grouped the sequences into 10 broadly defined clusters (I–X) based on evolutionary distance (Fig. 3). Within these clusters, sequences were closely affiliated with other environmental nirS clones retrieved from the sediments of Jiaozhou Bay (EU048441) [27], Bohai Gulf (JN257765), Bahía del Tóbari Estuary (KC614304) [28], Changjiang Estuary (EU235754; EU235973), Chesapeake Bay Estuary (DQ675714; DQ676127; DQ676123) [6], East Lake (HQ427998), Pearl River Estuary (HM773302), coastal and deep sea regions (DQ159648; GU348416) [29], as well as clones obtained from activated sludge (GU564887) [30] and the cultivated denitrifier Dechloromonas sp. (AM230913) [31].

Neighbor-joining phylogenetic tree of nirS sequences derived from the intertidal marshes of the Yangtze Estuary, with the nirN gene from Pseudomonas aeruginosa PAO1 (accession no. D84475) used as an outgroup. OTUs here were defined at 25 % nucleotide acid divergence for the phylogenetic tree construction. Bootstrap values greater than 50 % of 1,000 resamplings are shown near nodes. The scale indicates the number of nucleotide substitutions per site. GenBank accession numbers are shown for sequences from other studies. Numbers in parentheses following each site name indicate the number of sequences recovered from each sampling site in summer (red) and winter (blue)
In the sediment of the Yangtze Estuary, the nirS sequences in Cluster I were the dominant type at the higher salinity (10 to 20 ppt: YY, LC, and winter DXG) habitats, accounting for 41.8 to 79.1 % of the nirS-type denitrifier communities. In the lower salinity (0 to 1.5 ppt: XP, LHK, WSK, and BLG) sites, however, the nirS-type denitrifier communities were co-dominated by cluster III and VII (accounting for 46.3 to 78.2 %; Figs. 3 and 4). In addition, the nirS sequences in Clusters VI were exclusively obtained from the lower salinity stations, and apparently correspond to “low-salinity” groups of estuarine denitrifiers (Fig. 3 and Supplementary Table S3). However, clusterV harbored the most omnipresent nirS-type denitrifiers of the Yangtze Estuary which occurred in all the 14 constructed clone libraries (accounting for 1.3 to 16.1 %; Supplementary Table S3). In this study, no apparent season-specific cluster (except cluster IX) was found throughout the whole phylogenetic tree (Fig. 3).

The community compositions and distributions of nirS-harboring denitrifiers in intertidal sediments of the Yangtze Estuary. Higher salinity sites (10 to 20 ppt): YY, LC, and winter DXG. Lower salinity sites (0 to 2.3 ppt): XP, LHK, WSK, BLG, and summer DXG
Distribution of nirS-Harboring Denitrifiers
Spatiotemporal distribution of nirS-harboring denitrifiers was investigated using weighed UniFrac PCoA analysis (Fig. 5). The first two PCoA principal coordinates (P1 and P2) explained 61.48 % of the community changes of nirS-harboring denitrifiers among all the sampling sites. The denitrifier assemblages at the study area were divided into two distinctive groups. The nirS-encoding bacterial communities in group I were recovered in the lower salinity sediments (XP, LHK, WSK, BLG, and summer DXG) whereas those in group II were retrieved in the higher salinity sediments (YY, LC, and winter DXG). Furthermore, the observed variations in the community composition were statistically compared using the UniFrac distance matrix (Fig. 6). Results showed that the nirS clone library obtained from the lowest salinity site (winter LHK) was significantly different from the clone libraries obtained from the highest salinity sites (summer YY and winter YY) (P < 0.05). Also, the predominant groups between the lower and higher salinity sites were different based on the phylogenetic analysis (Figs. 3 and 4). However, based on the distance matrix, as well as the UniFrac PCoA analysis, nirS gene libraries at all 7 sampling sites were statistically indistinguishable between summer and winter (P > 0.05; Figs. 5 and 6). This lack of statistically significant differences between summer and winter libraries suggests that nirS-harboring denitrifier community compositions were relatively stable in the sediments of the Yangtze Estuary.

The UniFrac weighed PCoA analysis of the nirS-harboring denitrifier communities. The first two principal coordinate axes (PC1 and PC2) are shown. Red circle and blue square symbols represent samples from the lower salinity and higher salinity sites, respectively. The summer samples are in red font while the winter samples are in blue. Higher salinity sites (10 to 20 ppt): YY, LC, and winter DXG. Lower salinity sites (0 to 2.3 ppt): XP, LHK, WSK, BLG, and summer DXG

The UniFrac Distance Matrix of nirS gene clone libraries. The summer samples are in red font while the winter samples are in blue. The values show the UniFrac distances between each pair of clone libraries. Boldface blue data indicate that the libraries are drawn from significantly different communities (P < 0.05). All P values have been corrected for multiple comparisons by multiplying the calculated P value by the number of comparisons made (Bonferroni correction)
Quantification Analysis of nirS Gene
In this study, the standard curve spanned a range from 1.37 × 104 to 1.37 × 109 copies per microliter. Real-time qPCR consistency was confirmed by the strong linear inverse relationship between the threshold cycle (C T ) and the log10 value of nirS copy number of the standard curve (R 2 = 0.9959), with an amplification efficiency of 97.1 %. Melting curve analyses showed only one observable peak at 88.47 °C, while no detectable peaks associated with primer–dimer artifacts or other non-specific PCR amplification products were observed, confirming that fluorescent signals were derived from specific PCR products.
qPCR results showed great heterogeneous distribution of the nirS gene abundance among the sampling sites (Fig. 7). The highest number of nirS gene copies (9.00 × 107 copies per gram dry sediment) was observed at site XP in winter, whereas the lowest gene copy number (1.01 × 106 copies per gram dry sediment) was recorded at site LC in summer. The abundance of nirS-harboring denitrifiers was significantly higher at the lower salinity sites (6.37 × 106 to 9.00 × 107 copies per gram dry sediment) than at the higher salinity sites (1.01 × 106 to 7.50 × 106 copies per gram dry sediment; P < 0.05). Overall, no significant seasonal variation of nirS gene abundance was observed in intertidal sediments of the Yangtze Estuary (Student’s t test, P > 0.05; averaged 3.09 × 107 and 2.49 × 107 copies per gram dry sediment in summer and winter respectively), with the only exception of site BLG where the nirS gene abundance was 4.5 times higher in summer than in winter.

The spatiotemporal variations of nirS gene abundance in the marsh sediments of the Yangtze Estuary. Vertical bars indicate standard error (n = 3)
Relationships of nirS-harboring denitrifier communities and abundance with environmental variables
The potential relationships between the nirS-harboring denitrifier communities and environmental factors in the estuarine marshes were examined by the weighted CCA analysis (Fig. 8). The first two CCA axes (CCA1 and CCA2) explained 27.0 % of the total variance in the nirS-harboring denitrifier composition and 40.6 % of the cumulative variance of the genotype-environment relationship. The results indicated that the nirS-harboring bacterial community structures in the sediments of the Yangtze Estuary correlated significantly with salinity (P = 0.002, F = 2.37, 499 Monte Carlo permutations), ammonium-N (P = 0.004, F = 1.65, 499 Monte Carlo permutations), and nitrate-N (P = 0.049, F = 1.48, 499 Monte Carlo permutations). These factors had significant correlation with the composition and distribution of the communities, and provided 32.0 % of the total CCA explanatory power. Although the contribution of all other measured environmental factors (temperature, grain size, nitrite-N, organic carbon, and total phosphorus) was not statistically significant (P > 0.05, 499 Monte Carlo permutations), the combination of these variables provided additionally 34.5 % of the total CCA explanatory power. Of all the environmental factors investigated, only salinity showed a significant correlation with the nirS gene diversity (r = −0.549, P = 0.042, n = 14). We also investigated the correlations of nirS gene abundance with environmental variables. Pearson correlation analyses revealed that the nirS gene abundance was significantly related to the change of salinity (r = −0.589, P = 0.027, n = 14), as compared with other environmental factors.

CCA ordination plots for the first two principal dimensions of the relationship between nirS-harboring denitrifier communities with the environmental parameters. The summer samples are in red font and the winter samples are in blue. Green circle and pink triangle symbols represent samples from the lower salinity and higher salinity sites, respectively. Temp, NH4-N, NO3-N, NO2-N, TP, OC, and MΦ represent temperature, ammonium, nitrate, nitrite, total phosphorus, organic carbon, and sediment mean size, respectively
Discussion
In the present study, spatial and seasonal variations of nirS-harboring bacterial community structure, biodiversity, and abundance were measured in the intertidal sediments of the Yangtze Estuary to provide deeper insights into the microbial mechanisms driving denitrification in the estuarine environments. Diversity estimators of the nirS gene libraries in this study were within the same range previously reported at other environmental ecosystems [6, 27, 29, 32–35]. However, the nirS gene diversity at the study area was significantly higher than that of the bacterial and archaeal amoA genes, and anammox bacterial 16S rRNA gene (Student’s t test, P = 0.000) [8, 15], showing that a greater variety of microorganisms were involved in the denitrification process in the estuarine ecosystems [36]. Nevertheless, the dominant nirS genotypes observed here are not obviously affiliated with known denitrifying strains, which implies that we know very little about the group of organisms that are numerically dominant and ubiquitous in the estuarine systems.
The community of nirS-harboring denitrifiers at the study area showed distinctive spatial heterogeneity along the estuary (Figs. 4 and 5). At the lower salinity sites, the nirS-harboring bacterial community was co-dominated by cluster III and VII which on average occupied 62.8 % of the total detected sequences. At the higher salinity sites, however, it was dominated by cluster I which on average accounted for 59.7 % of the sequences. These results indicated that salinity was a key environmental factor regulating the biogeographical distribution of the nirS-type denitrifer community structure in the Yangtze Estuary. This trend was consistent with the previous results in other estuarine sediments [6] and was also consistent with a study reporting that nirS diversity was inversely correlated with salinity in a wastewater treatment plant [37]. Furthermore, this distribution pattern was supported by the CCA analysis in this study (Fig. 8). Interestingly, according to our previous study, the same distribution pattern was also observed for the anammox bacterial communities [8] and ammonia-oxidizing prokaryotic communities [15] in the sediments of the Yangtze Estuary, implying that salinity might play a critical role in the whole N cycle in the estuarine environments.
While salinity has a direct, although imperfectly understood, effect on the observed distribution patterns of nirS-harboring bacterial assemblages, it is worth noting that a variety of physical/chemical environmental parameters might also be important in structuring estuarine denitrifiers with complex interactions [6, 27]. In this study, the communities and spatial distributions of nirS-harboring denitrifiers also had significant correlations with ammonium and nitrate concentrations. Consistent with our results, it has been reported that ammonium contributed significantly to the denitrifier–environment relationship in the sediments of Jiaozhou Bay [27]. Nitrate supply is known to have a strong impact on denitrification rates, and might also have great impact on defining denitrifier community structures [6, 38]. Also, the impact of nitrate on the distribution of nirS-containing denitrifiers has been proposed in sediments within an oxygen-deficient zone off the Pacific coast [39]. Grain size of sediments might also have significant influence on nirS-containing bacterial communities, because it might control many physicochemical characteristics of sediment as it was related to in situ hydrological conditions, such as river runoff, tides, water mixing, and the intensity and dynamics of these activities [40]. However, no significant correlation between grain size and nirS-type denitrifier communities was observed in this study. Additionally, organic carbon, the primary electron donor for denitrifying bacteria, was previously reported to contribute significantly to the denitrifying bacteria–environment relationship, which was not confirmed in this study [27, 34]. However, the nirS-encoding denitrifier communities at the sampling sites did not show apparent seasonal shifts between summer and winter, suggesting that their population distribution in the Yangtze Estuary reflects an adaptation to site-specific characteristics.
The numbers of nirS-containing bacteria quantified in the marsh sediments of the Yangtze Estuary were comparable to those measured in the sediments of Bahía del Tóbari estuary (2.72 × 106 to 8.82 × 107 copies per gram sediment) [28] and San Francisco Bay estuary (5.4 × 105 to 5.4 × 107 copies per gram sediment) [41], and were relatively higher than those measured in deep-sea subsurface sediments of the South China Sea (4.29 × 105 to 2.06 × 106 copies per gram sediment) [34] and paddy field soil (6.0 × 105 to 1.2 × 106 copies per gram soil) [42]. Dong et al. observed a decline in the abundance of nirS-type denitrifiers in sediments as nitrate concentration decreased along the hyper-eutrophic Colne estuary (United Kingdom) [43]. However, we did not detect any significant correlations between nirS gene abundance and the environmental factors (temperature, ammonium, nitrate, nitrite, total phosphorus, organic carbon, and sediment mean size) analyzed in this study, with the only exception of salinity. In the present study, we observed that nirS gene abundance was significantly affected by salinity, and this result further confirmed that salinity might play a vital role in the N cycle of the estuarine ecosystems. Overall, the abundance of nirS-encoding denitrifiers did not show significant seasonal variations in this study, indicating that the number of the nirS type denitrifiers was not sensitive to the change of temperature. This relationship might also suggest that their population distribution in the Yangtze Estuary tend to show an adaptation to site-specific features.
In the intertidal sediments of the Yangtze Estuary, nirS-encoding nitrite-reducing bacteria had significantly higher abundance than anammox bacteria (anammox bacterial 16S rRNA gene abundance: 2.63 × 106 to 1.56 × 107 copies per gram sediment) [8] (Student’s t test, P = 0.023), indicating the nirS-encoding bacteria might play a more important role on nitrite reduction than anammox bacteria. Actually, it was estimated that denitrification contributed 87.1 % to 93.4 % to the total nitrogen loss from the intertidal sediments of the Yangtze Estuary based on the 15 N tracing experiments [8]. In addition, the abundance of nirS gene was significantly higher than that of amoA genes (bacterial amoA gene abundance: 7.36 × 104 to 5.20 × 106 copies per gram sediment; archaeal amoA gene abundance: 5.70 × 104 to 6.84 × 105 copies per gram sediment) in the Yangtze Estuary [15], which supported the hypothesis described by Corredor et al. [44]: at the estuarine level, ammonia oxidizers are less abundant than denitrifiers. The abundance of denitrifiers may be reflected by the denitrification rates. However, in this study we did not observe any significant correlations between nirS gene abundance and denitrification rates (P > 0.05). We have to note that this lack of correlation might be explained by the fact that we only targeted nirS-encoding type of the denitrifying abundance.
In summary, we explored the diversity, abundance, and distribution of cytochrome cd1-type nitrite reductase (nirS) sequences in intertidal sediments of the Yangtze Estuary. Generally, nirS-encoding bacterial biodiversity was significantly higher at the lower salinity sites than at the higher salinity sites, while no obvious seasonal variations were observed. The composition of nirS-harboring denitrifier communities at the study area showed distinctive spatial heterogeneity along the estuary, which were significantly correlated with salinity, ammonium, and nitrate. The nirS gene abundance varied between 1.01 × 106 and 9.0 × 107 copies per gram dry sediment, which were significantly related to salinity. However, no significant seasonal variation in the nirS gene abundance was observed at the study area. The present study showed the dynamics of nirS-harboring denitrifiers, and thereby, provided an opportunity to further understand the microbial mechanisms of the denitrification process.
References
Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai ZC, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA (2008) Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320:889–892
Seitzinger SP (2008) Nitrogen cycle: out of reach. Nature 452:162–163
Seitzinger S, Harrison JA, Bohlke JK, Bouwman AF, Lowrance R, Peterson B, Tobias C, Van Drecht G (2006) Denitrification across landscapes and waterscapes: a synthesis. Ecol Appl 16:2064–2090
Caffrey JM, Bano N, Kalanetra K, Hollibaugh JT (2007) Ammonia oxidation and ammonia-oxidizing bacteria and archaea from estuaries with differing histories of hypoxia. ISME J 1:660–662
Gruber N, Galloway JN (2008) An Earth-system perspective of the global nitrogen cycle. Nature 451:293–296
Francis CA, O’Mullan GD, Cornwell JC, Ward BB (2013) Transitions in nirS-type denitrifier diversity, community composition, and biogeochemical activity along the Chesapeake Bay estuary. Front Microbiol 4:237
Rich JJ, Dale OR, Song BK, Ward BB (2008) Anaerobic ammonium oxidation (anammox) in Chesapeake Bay sediments. Microb Ecol 55:311–320
Hou LJ, Zheng YL, Liu M, Gong J, Zhang XL, Yin GY, You L (2013) Anaerobic ammonium oxidation (anammox) bacterial diversity, abundance, and activity in marsh sediments of the Yangtze Estuary. J Geophys Res Biogeosci 118:1237–1246
Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616
Nogales B, Timmis KN, Nedwell DB, Osborn AM (2002) Detection and diversity of expressed denitrification genes in estuarine sediments after reverse transcription-PCR amplification from mRNA. Appl Environ Microbiol 68:5017–5025
Abell GCJ, Revill AT, Smith C, Bissett AP, Volkman JK, Robert SS (2010) Archaeal ammonia oxidizers and nirS-type denitrifiers dominate sediment nitrifying and denitrifying populations in a subtropical macrotidal estuary. ISME J 4:286–300
Zhu JR, Wang JH, Shen HT, Wu H (2005) Observation and analysis of the diluted water and red tide in the sea off the Changjiang River mouth in middle and late June 2003. Chin Sci Bull 50:240–247
Liu SM, Zhang J, Chen HT, Wu Y, Xiong H, Zhang EF (2003) Nutrients in the Changjiang and its tributaries. Biogeochemistry 62:1–18
Chai C, Yu ZM, Song XX, Gao XH (2006) The status and characteristics of eutrophication in the Yangtze River (Changjiang) Estuary and the adjacent East China Sea, China. Hydrobiologia 563:313–328
Zheng YL, Hou LJ, Newell S, Liu M, Zhou JL, Zhao H, You LL, Cheng XL (2014) Community dynamics and activity of ammonia-oxidizing prokaryotes in intertidal sediments of the Yangtze Estuary. Appl Environ Microbiol 80:408–419
Braker G, Fesefeldt A, Witzel KP (1998) Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl Environ Microbiol 64:3769–3775
Maidak BL, Cole JR, Parker CT Jr, Garrity GM, Larsen N, Li B, Lilburn TG, McCaughey MJ, Olsen MJ, Overbeek R, Pramanik S, Schmidt TM, Tiedje JM, Woese CR (1999) A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res 27:171–173
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Throbäck IN, Enwall K, Jarvis Å, Hallin S (2004) Reassessing PCR primers targeting nirS, nirK, and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49:401–417
Mohamed NM, Saito K, Tal Y, Hill RT (2010) Diversity of aerobic and anaerobic ammonia-oxidizing bacteria in marine sponges. ISME J 4:38–48
ter Braak CJF, šmilauer P (2002) CANOCO reference manual and CanoDraw for Windows User’s Guide: software for canonical community ordination (version 4.5). Microcomputer Power, Ithaca, NY
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5:169–172
Dang H, Wang C, Li J, Li T, Tian F, Jin W, Ding Y, Zhang Z (2009) Diversity and distribution of sediment NirS-Encoding bacterial assemblages in response to environmental gradients in the Eutrophied Jiaozhou Bay, China. Microb Ecol 58:161–169
Beman JM (2013) Activity, abundance, and diversity of nitrifying archaea and denitrifying bacteria in sediments of a subtropical estuary: Bahía del Tóbari, Mexico. Estuar Coast. doi:10.1007/s12237-013-9716-y
Santoro AE, Boehm AB, Francis CA (2006) Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Appl Environ Microbiol 72:2102–2109
Zafiriadis I, Ntougias S, Nikolaidis C, Kapagiannidis AG, Aivasidis A (2011) Denitrifying polyphosphate accumulating organisms population and nitrite reductase gene diversity shift in a DEPHANOX-type activated sludge system fed with municipal wastewater. J Biosci Bioeng 111:185–192
Heylen K, Gevers D, Vanpary B, Wittebolle L, Geets J, Boon N, De Vos P (2006) The incidence of nirS and nirK and their genetic heterogeneity in cultivated denitrifiers. Environ Microbiol 8:2012–2021
Yan T, Fields MW, Wu L, Zu Y, Tiedje JM, Zhou J (2003) Molecular diversity and characterization of nitrite reductase gene fragments (nirK and nirS) from nitrate- and uranium-contaminated groundwater. Environ Microbiol 5:13–24
Novinscaka A, Goyerb C, Dandieb CE, Filiona M (2013) Abundance, diversity and spatio-temporal dynamics of nirS gene-harbouring denitrifiers in a potato field over the course of a growth season. Syst Appl Microbiol 36:112–115
Li M, Hong Y, Cao H, Klotz MG, Gu JD (2013) Diversity, abundance, and distribution of NO-forming nitrite reductase-encoding genes in deep-sea subsurface sediments of the South China Sea. Geobiology 11:170–179
Guo L, Hu Z, Fang F, Liu T, Chuai X, Yang L (2014) Trophic status determines the nirS-denitrifier community in shallow freshwater lakes. Ann Microbiol 64:999–1006
Jetten MSM (2008) The microbial nitrogen cycle. Environ Microbiol 10:2903–2909
Yoshie S, Noda N, Tsuneda S, Hirata A, Inamori Y (2004) Salinity decreases nitrite reductase gene diversity in denitrifying bacteria of wastewater treatment systems. Appl Environ Microbiol 70:3152–3157
Wallenstein MD, Myrold DD, Firestone M, Voytek M (2006) Environmental controls on denitrifying communities and denitrification rates: insights from molecular methods. Ecol Appl 16:2143–2152
Liu X, Tiquia SM, Holguin G, Wu L, Nold SC, Devol AH, Luo K, Palumbo AV, Tiedje JM, Zhou J (2003) Molecular diversity of denitrifying genes in continental margin sediments within the oxygen-deficient zone off the pacific coast of Mexico. Appl Environ Microbiol 69:3549–3560
Dang H, Li J, Chen R, Wang L, Guo L, Zhang Z, Klotz MG (2010) Diversity, abundance, and spatial distribution of sediment ammonia-oxidizing Betaproteobacteria in response to environmental gradients and coastal eutrophication in Jiaozhou Bay, China. Appl Environ Microbiol 76:4691–4702
Mosier AC, Francis CA (2010) Denitrifier abundance and activity across the San Francisco Bay estuary. Environ Microbiol 2:667–676
Yoshida M, Ishii S, Otsuka S, Senoo K (2009) Temporal shifts in diversity and quantity of nirS and nirK in a rice paddy field soil. Soil Biol Biochem 41:2044–2051
Dong LF, Smith CJ, Papaspyrou S, Stott A, Osborn AM, Nedwell DB (2009) Changes in benthic denitrification, nitrate ammonification, and anammox process rates and nitrate and nitrite reductase gene abundances along an estuarine nutrient gradient (the Colne Estuary, United Kingdom). Appl Environ Microbiol 75:3171–3179
Corredor JE, Howarth RW, Twilley RR, Morell JM (1999) Nitrogen cycling and anthropogenic impact in the tropical interAmerican seas. Biogeochemistry 46:163–178
Acknowledgments
This work was funded by the National Natural Science Foundations of China (Nos. 41130525, 41322002, 41071135, 41271114 and 41021064), the Academic Doctoral Prize of East China Normal University (XRZZ2013024), the State Key Laboratory of Estuarine and Coastal Research (SKLEC, No. 2010RCDW07), and the SKLEC Fostering Project for Top Doctoral Dissertations. It was also supported by Program for New Century Excellent Talents in University (NCET) and the Marine Scientific Research Project for Public Interest (No. 200905007). The anonymous reviewers are thanked for their constructive comments.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yanling Zheng, Lijun Hou, and Min Liu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zheng, Y., Hou, L., Liu, M. et al. Diversity, Abundance, and Distribution of nirS-Harboring Denitrifiers in Intertidal Sediments of the Yangtze Estuary. Microb Ecol 70, 30–40 (2015). https://doi.org/10.1007/s00248-015-0567-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0567-x