
Abstract
The impact of fertilization regimes on sequential denitrifying communities was investigated in a rice paddy field with 17 years continuous fertilization, located in Taoyuan Agro-ecosystem Research Station (110°72″ E, 28°52″ N), China. The diversity, community composition, and size of denitrifying genes of narG, qnorB, and nosZ were determined using molecular tools including terminal restriction fragment length polymorphism, quantitative polymerase chain reaction (qPCR), cloning, and sequencing analysis. Soil samples were collected from the plots with no fertilizer (NF), urea (UR), balanced mineral fertilizers (BM), and BM combined with rice straw (BMR). UR and BM caused marked increase in the community size of the denitrifying genes; however, BMR resulted in the highest abundance. The community size of narG was the most affected by the fertilization regimes, while qnorB was the least. Fertilization also induced some shifts in the composition of denitrifying genes, but the responses of different genes varied. However, fertilization regimes caused no significant changes to the diversity of the denitrifying genes. Potential denitrification activity (PDA) was significantly correlated with the abundance of narG and nosZ rather than qnorB, but there were no such correlations between PDA and the composition and diversity of denitrifying communities. Conclusively, long-term fertilization significantly affected denitrifying community size and composition, but not diversity. Among the sequential denitrifying genes, narG was the most, while qnorB was the least sensitive communities to fertilization regimes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Denitrification is a complex microbial dissimilatory process in which nitrate and nitrite are reduced to gaseous NO, N2O, and N2 when oxygen is limited. Denitrification not only causes nitrogen loss from agricultural soil resulting in a low rate of nitrogen usage by crops but also contributes to greenhouse gas N2O emission. This can induce destruction of the stratospheric ozone layer, and its global warming potential is about 296 times higher than that of CO2 [1]. A wide range of taxonomic groups, including Bacteria, Archaea, and Fungi, have the capability to carry out denitrification; for instance, more than 50 genera of bacteria have been identified with denitrifying function [2]. Because of the high phylogenetic diversity among denitrifiers, 16S rRNA gene-based approaches are not suitable to explore the denitrifying communities [2]. Instead, the use of functional genes involved in denitrification is a more direct method of detecting or identifying denitrifying bacteria [3]. Complete denitrification requires the sequential action of four enzymes, nitrate reductase (encoded by narG and napA), nitrite reductase (encoded by nirK/nirS), nitric oxide reductase (encoded by nor), and nitrous oxide reductase (encoded by nosZ).
In the past decades, chemical fertilization has greatly contributed to food production, and the input is increasing continuously. For example, worldwide synthetic nitrogen fertilizer consumption was about 85 Tg N in 1999 with about 56 Tg N consumed in developing countries, and global N fertilizer consumption is projected to reach 105 Tg N by 2030 [4]. The heavy input of mineral nitrogen has had serious and long-term environmental consequences [5]. Nitrogen fertilizers can promote denitrification activity in agricultural soils; subsequently, substantial amounts of applied nitrogen fertilizer are lost through the denitrification process [6–8]. Denitrification rates are influenced by soil moisture and temperature, nutrient supply as nitrate and soluble organic matter, and soil texture [9, 10]. Spatial and temporal variations of these parameters and their interactions are often linked to the high variability of soil denitrification rates [11, 12]. Although previous studies have revealed that the above soil physical and chemical conditions indirectly affect denitrification, microbial factors are of great importance for directly regulating the denitrification process. Therefore, efforts should be taken to understand nitrogen turnover processes and the functional microorganisms involved.
Literature reviews have suggested that the composition and diversity of soil denitrifier communities may be the main microbial factors affecting denitrification [13, 14]. However, Hallin et al. [15] found that the community size correlated with the denitrifying process rate. This indicates that the soil denitrifier community may differ in response to environmental conditions that indirectly control the rate of denitrification [16, 17]. Recently, we reported the long-term fertilization effects on the nitrite reductase gene (nirK and nirS) community and revealed that the nirK community was more sensitive to fertilization than the nirS community. These findings guide us to explore the complete picture of key genes involved in denitrification in paddy soil because denitrification should be considered a sequential community process. Since not all denitrifiers produce the full suite of enzymes to complete the process, it remains unclear which are the main microbial groups involved in each denitrifying process and which community’s shifts are related to the potential denitrifying activity in paddy soil. However, most previous studies on soil denitrifiers have focused on a single or a couple denitrifying genes, rather than the sequential functional genes of the entire denitrification process [18–21]. Thus, it is worth detecting and comparing the complete denitrifying communities of denitrification. In addition, most previous studies on denitrifiers have focused on upland soils; few have been conducted on paddy soil, which is important agricultural land, especially in Asian countries [22]. Unlike upland soils, frequently flooded paddy soil supplies an anaerobic condition favoring denitrification [23]. However, knowledge of the denitrifying communities in paddy soils is very limited.
The main objectives of this study were to determine the long-term effects of repeated application of mineral fertilizers and rice straw incorporation on the composition, diversity, and abundance of sequential denitrifying communities and to explore the response of each denitrifying gene community to fertilization regimes in the paddy soil.
Materials and methods
Soil samples
The same soil samples were used in this study as the previous work, and the main characteristics have been described previously [24]. Briefly, soil samples were collected from the Taoyuan Agro-ecosystem Research Station of the Chinese Academy of Sciences (110°72″ E, 28°52″ N), China. A long-term experiment was established in 1990, including three replicates of ten treatments. Four treatments were selected for this study as follows: NF (no fertilizer), UR (182.3 kg N ha−1 a−1 as urea), BM (balanced mineral fertilizer, 182.3 kg N ha−1 a−1, 39.3 kg P ha−1 a−1, 197.2 kg K ha−1 a−1 as urea, superphosphate, and potassium chloride, respectively) and BMR (BM combined with rice straw produced in the plot). Soil samples were collected in March 2007, 10 months after the latest fertilizer application.
Potential denitrifying activity
Potential denitrifying activity (PDA) was performed according to the C2H2 inhibition method [18, 25]. Briefly, 25 g fresh soil were placed in 250-ml flasks containing 25 ml substrate with 1 mM glucose and 1 mM KNO3. Denitrifying conditions were achieved by evacuating and filling flasks with nitrogen gas three times. Acetylene was injected to a partial pressure of 10 kPa to inhibit the N2O reductase. The soils were incubated at 25°C on a rotary shaker at 210 rpm for 6 h. Head space samples were collected every hour. Nitrous oxide was analyzed with a gas chromatograph (Hewlett 5890; Packard series II) equipped with a poraplot Q column (3 m × 2 mm) and a 63Ni electron capture detector.
Soil microbial DNA preparation
Soil microbial DNA was extracted according to the previous method [24]. To minimize the influence of PCR inhibitors, extracted DNA was further purified using the Agarose Gel DNA Purification Kit (Takara Bio Inc, Japan). DNA concentration and quality were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Primers for denitrifying gene fragments
Degenerate primers for narG and nosZ genes were newly designed (Table 1). In total, 14 narG sequences and 15 nosZ sequences from various families or strains derived mostly from soils (Supplementary Table S1) were retrieved from the GenBank databases and aligned using ClustalW2 software (http://www.ebi.ac.uk/clustalw). Conserved peptide fragments were used to design forward and reverse primers (Table 1). The primer pairs of narG145F/773R and nosZ1126F/1884R were used for terminal restriction fragment length polymorphism (T-RFLP). While the primers of narG571F/773R and nosZ1126qF/1381R were used for real-time PCR. The specificity of the newly designed primers was validated with 30 strains, which were obtained from different sources (Supplementary Table S2).
Amplification of qnorB gene fragments was conducted using the qnorB 2F/7R (T-RFLP) and 2F/5R (qPCR) [26] with a few modifications (Table 1). The qPCR for 16S rRNA was performed with 1369F/1492R primer set [27].
PCR amplification and T-RFLP detection
The PCR reaction solution consisted of about 60 ng of DNA template, 400 nM dNTP, 400 nM of each primer, 1.5 mM Mg2+ buffer, and 2 U Taq DNA polymerase (Tiangen, China), and dH2O to make a final volume of 50 μL. Touchdown PCR for narG and nosZ were performed with 5 min of denaturation at 95°C, followed by 35 cycles of 30 s at 94°C, 30 s at 55 to 52°C, and 45 s at 72°C. The first two cycles were at 55°C, the following three cycles were at 52°C, and the last 30 cycles were at 50°C. PCR was completed with 7 min at 72°C. The program for qnorB was as follows: following 5 min of denaturation at 95°C, 5 cycles at the annealing temperature of 60°C, then 55°C for 35 cycles; the other program steps were the same with the narG.
T-RFLP analysis was carried out by Sangni Corporation (Shanghai, China) using an ABI Prism 3100 Genetic Analyzer. T-RFLP profiles for narG were generated with endonucleases TaqI and RsaI, for qnorB with MspI and HhaI, and for nosZ with AluI and HhaI.
Cloning and sequencing
After the PCR amplification, the expected bands of narG, qnorB, and nosZ genes were excised from agarose gel and purified with Wizard SV Gel and PCR clean-up Systems (Promega, Madison, USA) following manufacturer’s instructions. The PCR products were cloned into a pGEM-T vector (Promega) and transformed into Escherichia coli strain DH-5α, followed by “blue-white screening.” Plasmids with correct size inserts were screened through PCR with M13F and M13R primers. For each of the above gene, about 85 selected colonies from each treatment were sequenced by ABI Prism 3100 Genetic Analyzer (Invitrogen Biotechnology Co., Guangzhou). The detailed number of sequences is shown in supplementary material (Table S4). All narG, qnorB, and nosZ gene sequences determined by the BLAST program in the present study have been deposited in GenBank under the accession numbers FJ209539–FJ209729 (narG), HQ615163–HQ615366 (qnorB), and FJ209365–FJ209538 (nosZ)
Quantitative PCR
The primer pairs of 16S rRNA-1369F/1492R, narG-571F/773R, and nosZ 1126qF/1318R qnorB-2F/5R were employed for qPCR of 16S rRNA, narG, nosZ, and qnorB genes, respectively (Table 1). The real-time PCR assay was carried out in a volume of 10 μL, and the assay mixture contained 0.25× SYBR green (Invitrogen, USA), 1 μL of 10× reaction buffer, 0.8 μL of 2.5 mmol L−1 dNTP, 3.5 mmol L−1 Mg2+, 0.5 U Hot Start DNA polymerase (Takara, Japan), 150 nmol L−1 forward and reverse primers, and 5 ng of template DNA. Thermal cycling conditions were as follows: an initial cycle of 95°C for 3 min; 5 cycles of 95°C for 15 s, 60°C for 20 s, and 72°C for 20 s; and 30 cycles of 95°C for 15 s, 55°C for 20 s, and 72°C for 20 s. qPCR was performed with an ABI Prism 7900 sequence detection system. The standard curves for 16S rRNA, narG, qnorB, and nosZ were created using 10-fold dilution series (102–109 copies, Supplementary Table S3 and Fig. S2) of the plasmids containing the targeted gene fragments from soil as described by Henry et al. [28]. The efficiency of the reactions was from 82% to 110% for the above genes (Supplementary Table S3). The R 2 values for the curves were above 0.99 (Table S3). One sharp peak was observed in each of the melt curves (Supplementary Fig. S3).
Statistical analysis
The size and relative abundance of terminal restriction fragments (T-RFs) were quantified using PeakScan version 1.0 software (Applied Biosystems, Inc.). The fragments with a signal above 1% of the sum of all peak heights were included. The peak heights of T-RFs that differed in size by ≤2 bp in an individual profile were summed and considered as one fragment. T-RFs with a size of more than 90 bp were used for cluster analysis. Diversity indices were calculated with PC-ORD version 5.0. Ordination techniques of correspondence analysis (CA) and canonical correspondence analysis (CCA) (CANOCO version 4.53) [29] were used for community analysis. CCA was used to assess the relationship between microbial community profiles and environmental variables. Sequence identity >95% was defined as an operational taxonomic unit (OTU) [30]. The clone sequences were aligned using ClustalX (1.83), and a neighbor-joining tree was produced from the MEGA 4.0 [31]. Bootstrap analysis was used to estimate the reliability of the phylogenetic reconstruction (1,000 replicates).
Results
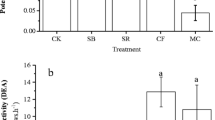
Potential denitrifying activity
Long-term fertilization affected PDA. PDA was significantly higher in UR, BM, and BMR treatments (220, 161, and 394 ng N2O–N g−1 dry soil h−1, respectively) than in NF (63 ng N2O–N g−1 dry soil h−1). There was no significant difference between the UR and the BM treatments, and the highest PDA was observed in plots amended with rice straw (BMR).
Primer validation and denitrifying gene amplification
Both narG and nosZ gene fragments were successfully amplified from all the 12 soil samples by the primer sets of narG-145F/773R and nosZ-1126F/1884R. In total, 344 and 331 clones generated by narG-145F/773R and nosZ-1126qF/1884R, respectively, were sequenced (about 85 clones for narG and nosZ, respectively, from each treatment, details shown in Supplementary Table S4). The consequent alignment analysis by BLAST indicated that 94% of narG clones and 88% of nosZ clones were homologous to narG and nosZ genes, respectively (data not shown).These high percentages implied that the newly designed primers possess high specificity for the denitrifying genes in the soil.
Similarly, a total of 318 clones were generated by the primer set of qnorB-2F/7R and resulted in 97% positive proportion (307 clones) that were homologous to the qnorB gene fragments in the GenBank database.
The nitric oxide reductase gene, cnorB, was also first in our consideration because this gene is directly linked to N2O production and is only found in denitrifiers, while the qnorB gene has been discovered both in denitrifiers and in some nondenitrifying bacteria [32]. Unfortunately, the cnorB gene failed to amplify with the published cnorB primers [26]. We also designed a set of cnorB primers and obtained the targeted size of PCR products; however, most of these products were not homogeneous to cnorB. Therefore, the cnorB gene was not used for further analysis.
Influence of fertilization on the abundance of denitrifying genes
The copy number of each denitrifying gene (narG, nirK/nirS, qnorB, and nosZ) varied similarly to the fertilization regimes (Table 2). The highest copy number was observed in the plots amended with BMR followed by the two mineral fertilization treatments (BM and UR), and the lowest occurred in NF treatment. Compared with the above denitrifying communities, interestingly, fertilization exhibited less effect on the abundance of 16S rRNA. The mineral fertilization treatments of UR and BM did not induce significant differences compared to NF, while only BMR resulted in doubled 16S rRNA copy numbers, which was significantly higher than NF (Table 2). In contrast, the impact of fertilization on the size of denitrifying genes was more obvious because the two mineral fertilization regimes (UR and BM), especially amended with BM, caused remarkable increases in the copy numbers of the denitrifying genes. However, plots amended with BMR induced the highest copy numbers of the denitrifying genes, which was three to nine times higher compared to NF. The results implied that the abundance of the denitrifying genes of narG, nirK/nirS, qnorB, and nosZ was more sensitive to fertilization than that of 16S rRNA. However, the abundance of the denitrifying genes was not evenly influenced by the long-term fertilization, where narG and nirS communities were most sensitively responsive to the fertilizations followed by nirK and nosZ; qnorB was the least sensitive community.
The order of the denitrifying genes abundance was as follows: narG > (nirK + nirS) > qnorB > nosZ. This trend coincidently agreed with the process order of denitrification. narG gene works on the first step to reduce NO −3 to NO −2 and possessed the highest copy number (3.2 × 108 in NF), while the nosZ gene is involved in the last step of denitrification and possessed the lowest copy number (3.1 × 106 in NF). This finding indicates clearly that the copy numbers of the key genes involved in the denitrification process in paddy soil decreased with the sequential reaction order.
Diversity variations of the denitrifying genes under fertilization regimes
The diversity variations of the denitrifying genes based on the T-RFLP results are presented in Table 3. Statistically, no significant differences in the diversity indices of narG, qnorB, and nosZ were detected among the fertilization treatments except that UR induced significantly lower diversity of qnorB than the other treatments, indicating weak effect of fertilization on the diversity of narG, qnorB, and nosZ gene communities after 17 years of continuous fertilization. Combined with our previous results of nirK and nirS gene diversity, it suggested that the diversity of denitrifying gene communities in paddy soil is not sensitive to fertilization.
Effect of fertilization on denitrifying community composition
The community compositions of narG, qnorB, and nosZ genes were determined by T-RFLP. The T-RF profiles of the three genes from triplicate samples of each treatment were similar, indicating that the results were reproducible and representative for the denitrifier communities in the soils (Supplementary Fig. S1). Therefore, the average relative abundances of the T-RFs for narG (TaqI), qnorB (HhaI) and nosZ (AluI) are presented in Fig. 1. It is obvious that the community composition of narG varied significantly under both mineral fertilizations (UR and BM) and rice straw incorporation (BMR), while the community composition of nosZ was only affected by the rice straw incorporation (BMR), which induced shifts of some dominant T-RFs. Comparatively, the community composition of qnorB was less affected by the fertilization regimes.

Average relative abundances of narG (a) T-RFs with endonuclease TaqI, qnorB (b) with HhaI, and nosZ (c) with AluI from long-term fertilization treatments. The relative abundance of T-RFs is given as a percentage of the total peak height. Fragment sizes within the graph indicate the sizes (bp) of the experimental T-RFs by T-RFLP
The relative abundance of narG T-RFs of 251 and 263 bp declined markedly in the plots amended with UR, BM, and BMR compared to the NF (Fig. 1a). However, the narG T-RFs of 275 and 437 bp were nearly undetectable in NF but dominant in UR, BM, and BMR. Obvious variations were also detected on narG T-RFs of 205 and 254 bp. The T-RFLP patterns of narG in BM and BMR were similar, although the relative abundance of some less-abundant fragments showed a little difference. Fewer variations in the relative abundance of qnorB and nosZ T-RFs were observed among the treatments (Fig. 1b, c) in comparison with narG. For the profiles of qnorB, no significant differences were detected on the relative abundance of several dominant T-RFs (93, 107, 187, and 242 bp) and less-dominant T-RFs (165, 442, and 463 bp), indicating that the qnorB patterns of the four treatments were similar. For the profiles of nosZ, it was obvious that NF, UR, and BM treatments shared similar T-RFs patterns characterized by a predominant T-RF of 273 bp. Application of BMR resulted in lower proportion of T-RF of 273 bp and higher proportion of T-RF of 141 bp compared to other treatments. In addition, BMR treatment also induced three dominant groups of T-RFs of 283, 286, and 316 bp, which were minority groups in NF, UR, and BM treatments.
Correspondence analysis (CA) was employed to determine the fertilization effects on the narG-, qnorB-, and nosZ-defined communities (Fig. 2). The narG community composition formed three distinct groups (Fig. 2a) among the four treatments with three replicates, in which BM and BMR grouped together while NF and UR were grouped independently, indicating that mineral fertilizations obviously altered narG communities rather than rice straw incorporation. qnorB and nosZ denitrifier communities were not separated like narG. The qnorB samples from the four treatments scattered around the center of the ordination plot, suggesting that fertilization had a weak effect on qnorB-denitrifier community composition (Fig. 2b). For nosZ, only the replicates of BMR grouped independently, and the rest of the treatments grouped together, implying rice straw incorporation caused obvious alteration of nosZ community composition, while mineral fertilizers did not result in severe changes (Fig. 2c).

CA analysis of T-RFLP profiles examining the effect of different fertilization regimes on the composition of narG (a), qnorB (b), and nosZ (c) denitrifier communities in long-term fertilization plots. The nraG, qnorB, and nosZ distributions were based on the relative abundance of T-RFs after restriction using TaqI, HhaI and AluI enzymes, respectively
The relationship between PDA and composition and abundance of denitrifying community
PDA was not coupled to the denitrifier community composition, as determined by CCA analysis. However, statistical calculation showed that PDA was significantly correlated with the copy numbers of narG, nirK, nirS, and nosZ gene (R 2 = 0.792, 0.811, 0.855, and 0.732 respectively, P < 0.01 by Pearson correlation test). No such correlation existed between PDA and qnorB copy number (R 2 = 0.352, P > 0.05).
Phylogenetic analysis
Of the total 322 narG clone sequences, 143 OTUs were identified from all the treatments and formed the phylogenetic tree together with a selected published narG reference sequences (Fig. 3). The tree can be divided into six main clusters and some subclusters based on the location of NarG from cultivated microorganisms. Cluster I contained 46% of the total clones, which were closely related to the narG of Alphaproteobacteria (~92% identical at the protein level), and the proportion of the clones from each treatment was similar (43–50%). Unlike cluster I, clusters II and V were related to Betaproteobacteria, and the clones were unevenly distributed in these two clusters in relation to the fertilization regimes. For example, in cluster V, the clones from BM accounted for 25% of the total, whereas it was only about 3% for NF. Clusters III and IV were related to Gammaproteobacteria, and only a few clones (2%) were from BMR in cluster IV, while no obvious differences in clone distribution were observed in cluster III. Interestingly, about 2% of the narG clones were similar to Gram-positive bacteria (Mycobacterium), which were located in cluster VI. These results indicate that paddy soil contains highly diversed narG-containing denitrifiers, and fertilization regimes intensively influenced narG phylogeny.

Phylogenetic tree of narG sequences cloned from long-term fertilization plots. Bootstrap values (>50%) are indicated at the branch points. GenBank accession number followed by treatment abbreviation in parentheses, and the numbers before each treatment abbreviation represent the respective number of clones. The scale bar represents a 5% estimated sequence divergence
nosZ phylogenetic tree containing 103 OTUs mainly grouped into seven clusters. Most of the nosZ OTUs from paddy soil were related to Alpha-, Beta- and Gammaproteobacteria, the first two groups accounting for 96% of the total OTUs (Fig. 4). Forty-five nosZ Alphaproteobacterial OTUs were distributed in clusters I and III, closely related to the nosZ gene of Bradyrhizobium (83–95% identical at the protein level). nosZ-containing Betaproteobacteria accounted for 54 OTUs and allocated in clusters II, V, and VI; among them, cluster V was unique in which not a single clone was isolated from BMR treatment. The nosZ-containing Gammaproteobacteria comprised a very small group of clones, which were mainly isolated from UR and BMR treatments (cluster IV) and related to Pseudomonas stutzeri strain A1501.


Phylogenetic tree of qnorB sequences from a paddy field in long-term fertilization. Bootstrap values (>50%) are indicated at the branch points. GenBank accession number followed by treatment abbreviation in parentheses, and the numbers before each treatment abbreviation represent the respective number of clones. The scale bar represents a 10% estimated sequence divergence
Unlike the features of narG and nosZ, qnorB phylogeny is incongruent with the 16S rRNA phylogeny at the family or genus level (Fig. 5). For example, cluster VII contained most Betaproteobacteria, but was intermingled with the sequences from Gammaproteobacteria; the species of Ralstonia genus branched in different clusters (clusters VII and VIII) and grouped with bacteria of other genus. Another feature of the qnorB phylogenetic tree was that more than half of the qnorB clones (56%) assembled with non-cultured species in clusters III and VI. The effect of fertilization on qnorB phylogeny was in congruence with the T-RFLP analysis, indicating that the fertilization impact was not obvious. For example, in the two major clusters (I and VI), the proportions of the clones obtained from the fertilization treatments were quite evenly distributed (27–30% in cluster I and 35–40% in cluster VI).

Phylogenetic tree of nosZ sequences from a paddy field in long-term fertilization. Bootstrap values (>50%) are indicated at the branch points. GenBank accession number followed by treatment abbreviation in parentheses, and the numbers before each treatment abbreviation represent the respective number of clones. The scale bar represents a 5% estimated sequence divergence
Discussion
PCR primers with high specificity and universality are a prerequisite for molecular analysis of microbial communities, since they ultimately determine what is detected in the environmental sample. In this study, the narG and nosZ primers were designed in our laboratory based on the sequences isolated mainly from soil samples, to facilitate the specific soil denitrifier community. The specificity and universality of the newly designed narG primers was verified by amplifying narG denitrifiers representing a spectrum of Proteobacteria and Gram-positive bacteria. The tested nosZ primers could successfully amplify the majority of the Proteobacteria except for Gram-positive bacteria and Firmicutes, indicating the specificity for nosZ of Gram-negative bacteria, which coincided with previous studies [33–35]. However, the primer set nosZ-1126F/1884R showed the advantage of amplifying nosZ-containing Alpha- and Betaproteobacteria even compared to some previous nosZ primers with bias toward Alpha- or Betaproteobacteria [20, 34, 36]. Furthermore, the newly designed narG-145F/773 and nosZ-1126F/1884R primers obtained 92% and 87% of positive clones, respectively, which covered more than ten genera of Alpha-, Beta-, and Gammproteobacteria and a large number of uncultured species, showing that amplification with the new primers could detect a wide range of the corresponding gene communities.
Denitrification is an important stepwise process in soil nitrogen turnover, which is closely related to fertilization. Soil microbial denitrifiers, the major organisms in driving denitrification, can be affected by fertilizers [15]. However, little is known about how fertilization regimes affect stepwise denitrifying communities. As the influence of fertilization on denitrifiers would be an accumulative consequence after continuous application of fertilizers to soil, short-term experiment might not reflect the shifts of the functional microorganisms. Miller et al. [37] found that neither crop residues nor nitrogen fertilizer alone or in combinations induced a measurable change in the abundance of the nosZ gene in the short term. A long-term paddy field experiment was employed in this study to investigate the impact of fertilization regimes on denitrifying communities. We found that the indices of the diversity of narG, qnorB, and nosZ genes were little affected by the fertilization treatments, although some shifts of denitrifier composition occurred. It might mean that paddy soil possesses a strong capacity in maintaining a stable divergence of denitrifiers under applying fertilizer application, including mineral fertilizers and rice straw incorporation [38, 39]. In contrast, fertilization regimes induced a significant increase in the community size of all the denitrifying genes. Furthermore, we found that this influence was more obvious on denitrifying communities than on 16S rRNA because the increase in the copy numbers of each denitrifying gene (narG, nirK/nirS, qnorB, and nosZ) in the plots amended with fertilizers was about ten times higher than that of 16S rRNA. It might suggest that denitrifying communities were sensitive groups to fertilization practices within bacteria society. This might be due to the input of nitrogen substrates stimulating nitrogen turnover bacteria, and other nutrients supply, including C, P, and K, might accelerate the growth and reproduction of these functional microorganisms [40].
We found that the abundance of the denitrifying genes were quite different, narG possessed the highest copy number followed by nirK/nirS and qnorB, and nosZ had the lowest copy numbers, and the abundance of narG was about 100 times higher than nosZ. Coincidently, this abundance order of the genes is consistent with the step order of denitrification. The mechanism of such a big difference in abundance existing among the denitrifying genes is not clear. This phenomenon might be related to the gene copies within the genomes and the highly distinct physiologies between narG- and nosZ-containing organisms. It has been reported that nirK, qnorB, and nosZ only have one copy per genome, but narG could have up to three copies [41]. Substrate concentration might also play an important role in the different abundance. Nitrate is the first substrate for denitrification under anaerobic condition, and it is the most abundant compared to nitrite, NO, and N2O. Considering denitrification, plant uptake, leaching, and gas emission, the concentrations of the denitrifying intermediates in soil decrease significantly followed by the reduction order [42]. These substrate differences could be closely related to the abundance of the corresponding genes. Besides, the nitrate reductase encoded by narG gene is also involved in other dissimilatory pathway for nitrate reduction [43], and thus, more narG-containing bacteria might be required. Furthermore, the nosZ gene has been found absent in nearly one third of denitrifiers that can harbor nir and nor genes [44], and the reduction of N2O to N2 catalyzed by nosZ contributes relatively little to the overall bioenergetic needs of denitrifying organisms [45]. In addition, it is known that the results of gene abundance obtained in PCR-based studies are influenced by the initial choice of primer set [34]. Thus, the coverage of denitrifier communities to be amplified might be different for each PCR primer.
Fertilization regimes exhibited various effects on the abundance of the denitrifiers. Among them, BMR induced the highest copy numbers of all the denitrifying genes, which were 1.7- to 2.9-fold higher than BM. Rice straw incorporation might be an important contributor to the high copy numbers of the genes due to the supplementation of carbon and other nutrients for the microbial communities, and the majority of denitrifiers are heterotrophs, which rely on organic matter [40, 46]. Kandeler et al. [47] also found that the highest correlation existed between the quantity of soil organic carbon and the number of denitrifying genes in a receding glacier foreland. However, applying organic fertilizer alone without chemical fertilizers did not significantly increase the copy number of denitrifying genes (narG, nir, and nosZ) [15]. In the present study, the balanced mineral fertilization (BM) also produced significant increases in the abundance of the denitrifying genes compared to NF. The BM treatment obviously improved rice growth and yield, and as a result, more plant residues (roots) and secretions left in the soil could stimulate the reproduction of denitrifiers. However, it is poorly understood whether applying nitrogen alone also significantly increases the abundance of narG, nirS, and nosZ in comparison with the NF treatment, since plant growth was similar between the two treatments. It could be hypothesized that some strains favoring N might flourish under continuous input of nitrogen fertilizer. However, high abundance of denitrifiers in the plots amended with fertilizers might link to high denitrification rate, causing more nitrogen loss through N2O and N2. This speculation can be supported by the fact that soil potential denitrification activity was significantly correlated with the copy numbers of denitrifying genes, which was found both in this study and other work [15, 24].
We also detected that different denitrifying community compositions responded differentially to fertilization regimes. Mineral fertilization regimes induced obvious shifts of narG gene community composition; however, the influence of rice straw incorporation was very limited. In contrast, only rice straw resulted in notable variations of nosZ community structure, whereas the composition of qnorB was little affected by the fertilization regimes. Since these functional genes mostly belonged to different bacterial strains, it might be deduced that narG community composition was more sensitive to mineral fertilizers rather than rice straw incorporation, while nosZ composition was more susceptible to the later. Previous work indicated that nirK/nirS communities differentially responded to fertilization [24, 48]. Therefore, it is obvious that different denitrifying gene communities possess differential responses to fertilization practice. Although we do not know much about how significant these differential changes can influence nitrogen cycling, it might be an important factor in determining nitrogen turnover rate.
In conclusion, this study demonstrates that the sequential denitrifying gene communities responded differently to fertilization regimes. Long-term fertilization significantly affected the denitrifying community size and the denitrifying community composition. However, fertilization regimes did not result in significant variations of the diversity of denitrifiers. Functional communities involved in nitrogen cycling provide good models in microbial ecology for studying the role of the size and structure of microbial communities in corresponding process rates and ecosystem functioning.
References
Lashof DA, Ahuja DR (1990) Relative contributions of greenhouse gas emissions to global warming. Nature 344:529–531
Zumft WG (1992) The denitrifying prokaryotes. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes. A handbook on the biology of bacteria: ecophysiology, isolation, identification, applications. Springer, New York, pp 554–582
Wolsing M, Prieme A (2004) Observation of high seasonal variation in community structure of denitrifying bacteria in arable soil receiving artificial fertilizer and cattle manure by determining T-RFLP of nir gene fragments. FEMS Microbiol Ecol 48:261–271
Mosier A, Wassmann R, Verchot L, King J, Palm C (2004) Methan and nitrogen oxide fluxes in tropical agricultural soils: sources, sinks and mechanisms. Environ Dev Sustain 6:11–49
Canfield DE, Glazer AN, Falkowski PG (2010) The evolution and future of earth's nitrogen cycle. Science 330:192–196
Kaiser EA, Kohrs K, Kucke M, Schnug E, Heinemeyer O, Munch JC (1998) Nitrous oxide release from arable soil: importance of N-fertilization, crops and temporal variation. Soil Biology & Biochemistry 30:1553–1563
Deklein CAM, Vanlogtestijn RSP (1994) Denitrification in the top-soil of managed grasslands in The Netherlands in relation to soil type and fertilizer level. Plant and Soil 163:33–44
Knowles R (1982) Denitrification. Microbiol Rev 46:43–70
Starr RC, Gillham RW (1993) Denitrification and organic-carbon availability in 2 aquifers. Ground Water 31:934–947
Groffman PM, Hanson GC, Kiviat E, Stevens G (1996) Variation in microbial biomass and activity in four different wetland types. Soil Science Society of America Journal 60:622–629
Groffman PM, Tiedje JM (1988) Denitrification hysteresis during wetting and drying cycles in soil. Soil Science Society of America Journal 52:1626–1629
Hill AR, Devito KJ, Campagnolo S, Sanmugadas K (2000) Subsurface denitrification in a forest riparian zone: Interactions between hydrology and supplies of nitrate and organic carbon. Biogeochemistry 51:193–223
Wallenstein MD, Myrold DD, Firestone M, Voytek M (2006) Environmental controls on denitrifying communities and denitrification rates: insights from molecular methods. Ecol Appl 16:2143–2152
Philippot L, Hallin S (2005) Finding the missing link between diversity and activity using denitrifying bacteria as a model functional community. Curr Opin Microbiol 8:234–239
Hallin S, Jones CM, Schloter M, Philippot L (2009) Relationship between N-cycling communities and ecosystem functioning in a 50-year-old fertilization experiment. Isme Journal 3:597–605
Cavigelli MA, Robertson GP (2000) The functional significance of denitrifier community composition in a terrestrial ecosystem. Ecology 81:1402–1414
Hwang SJ, Hanaki K (2000) Effects of oxygen concentration and moisture content of refuse on nitrification, denitrification and nitrous oxide production. Bioresour Technol 71:159–165
Dambreville C, Hallet S, Nguyen C, Morvan T, Germon JC, Philippot L (2006) Structure and activity of the denitrifying community in a maize-cropped field fertilized with composted pig manure or ammonium nitrate. FEMS Microbiol Ecol 56:119–131
Jayakumar DA, Francis CA, Naqvi SWA, Ward BB (2004) Diversity of nitrite reductase genes (nirS) in the denitrifying water column of the coastal Arabian Sea. Aquat Microb Ecol 34:69–78
Stres B, Mahne I, Avgustin G, Tiedje JM (2004) Nitrous oxide reductase (nosZ) gene fragments differ between native and cultivated Michigan soils. Appl Environ Microbiol 70:301–309
Cao YP, Green PG, Holden PA (2008) Microbial community composition and denitrifying enzyme activities in salt marsh sediments. Appl Environ Microbiol 74:7585–7595
FAO (2003) Selected indicators of food and agriculture development in Asia-pacific region (1992–2002). RAP publication, 23–24
Akiyama H, Yan XY, Yagi K (2006) Estimations of emission factors for fertilizer-induced direct N2O emissions from agricultural soils in Japan: summary of available data. Soil Science and Plant Nutrition 52:774–787
Chen Z, Luo XQ, Hu RG, Wu MN, Wu JS, Wei WX (2010) Impact of long-term fertilization on the composition of denitrifier communities based on nitrite reductase analyses in a paddy soil. Microb Ecol 60:850–861
Pell M, Stenberg B, Stenström J, Torstensson L (1996) Potential denitrification activity assay in soil–With or without chloramphenicol? Soil Biology and Biochemistry 28:393–398
Braker G, Tiedje JM (2003) Nitric oxide reductase (norB) genes from pure cultures and environmental samples. Appl Environ Microbiol 69:3476–3483
Suzuki MT, Taylor LT, DeLong EF (2000) Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol 66:4605–4614
Henry S, Baudoin E, Lopez-Gutierrez JC, Martin-Laurent F, Baumann A, Philippot L (2004) Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR. Journal of Microbiological Methods 59:327–335
Bremer C, Braker G, Matthies D, Reuter A, Engels C, Conrad R (2007) Impact of plant functional group, plant species, and sampling time on the composition of nirK-type denitrifier communities in soil. Appl Environ Microbiol 73:6876–6884
Yoshida M, Ishii S, Otsuka S, Senoo K (2009) Temporal shifts in diversity and quantity of nirS and nirK in a rice paddy field soil. Soil Biology & Biochemistry 41:2044–2051
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings in Bioinformatics 5:150–163
Hendriks J, Oubrie A, Castresana J, Urbani A, Gemeinhardt S, Saraste M (2000) Nitric oxide reductases in bacteria. Biochimica Et Biophysica Acta-Bioenergetics 1459:266–273
Roesch C, Mergel A, Bothe H (2002) Biodiversity of denitrifying and dinitrogen-fixing bacteria in an acid forest soil. Appl Environ Microbiol 68:3818–3829
Henry S, Bru D, Stres B, Hallet S, Philippot L (2006) Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl Environ Microbiol 72:5181–5189
Scala DJ, Kerkhof LJ (1998) Nitrous oxide reductase (nosZ) gene-specific PCR primers for detection of denitrifiers and three nosZ genes from marine sediments. FEMS Microbiol Lett 162:61–68
Ruiz-Rueda O, Hallin S, Baneras L (2009) Structure and function of denitrifying and nitrifying bacterial communities in relation to the plant species in a constructed wetland. FEMS Microbiol Ecol 67:308–319
Miller MN, Zebarth BJ, Dandie CE, Burton DL, Goyer C, Trevors JT (2008) Crop residue influence on denitrification, N2O emissions and denitrifier community abundance in soil. Soil Biology and Biochemistry 40:2553–2562
Ogilvie LA, Hirsch PR, Johnston AWB (2008) Bacterial diversity of the Broadbalk ‘classical’ winter wheat experiment in relation to long-term fertilizer inputs. Microb Ecol 56:525–537
Freitag TE, Chang L, Clegg CD, Prosser JI (2005) Influence of inorganic nitrogen management regime on the diversity of nitrite-oxidizing bacteria in agricultural grassland soils. Appl Environ Microbiol 71:8323–8334
Cheneby D, Bru D, Pascault N, Maron PA, Ranjard L, Philippot L (2010) Role of plant residues in determining temporal patterns of the activity, size, and structure of nitrate reducer communities in soil. Appl Environ Microbiol 76:7136–7143
Philippot L (2002) Denitrifying genes in bacterial and archaeal genomes. Biochimica Et Biophysica Acta-Gene Structure and Expression 1577:355–376
Yu KW, Struwe S, Kjoller A, Chen GX (2008) Denitrification rate determined by nitrate disappearance is higher than determined by nitrous oxide production with acetylene blockage. Ecol Eng 32:90–96
Blasco F, Guigliarelli B, Magalon A, Asso M, Giordano G, Rothery RA (2001) The coordination and function of the redox centres of the membrane-bound nitrate reductases. Cellular and Molecular Life Sciences 58:179–193
Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61:533–616
Jones CM, Stres B, Rosenquist M, Hallin S (2008) Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol Biol Evol 25:1955–1966
Parkin TB (1987) Soil microsites as a source of denitrification variability. Soil Science Society of America Journal 51:1194–1199
Kandeler E, Deiglmayr K, Tscherko D, Bru D, Philippot L (2006) Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl Environ Microbiol 72:5957–5962
Yoshida M, Ishii S, Otsuka S, Senoo K (2010) nirK-harboring denitrifiers are more responsive to denitrification-inducing conditions in rice paddy soil than nirS-harboring bacteria. Microbes and Environments 25:45–48
Acknowledgments
We thank Professor Min Lin for providing Pseudomonas stutzeri A1501 and Professor Ron McLaren for English improvements. This work was financially supported by KZCX2-YW-BR-01, KZCX2-YW-T07, NSFC 41090282 and 41071181.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 1470 kb)
Rights and permissions
About this article
Cite this article
Chen, Z., Liu, J., Wu, M. et al. Differentiated Response of Denitrifying Communities to Fertilization Regime in Paddy Soil. Microb Ecol 63, 446–459 (2012). https://doi.org/10.1007/s00248-011-9909-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-011-9909-5