
Abstract
Moso bamboo is fast-growing and negatively allelopathic to neighboring plants. However, there is little information on the effects of its establishment and expansion to adjacent forest soil communities. To better understand the impacts of bamboo invasion on soil communities, the phylogenetic structure and diversity of the soil bacterial communities in moso bamboo forest, adjacent Japanese cedar plantation, and bamboo-invaded transition zone were examined using a combination of 16S rRNA gene clone libraries and bar-coded pyrosequencing techniques. Based on the number of operational taxonomic units (OTUs), Shannon diversity index, Chao1 estimator, and rarefaction analysis of both techniques, the bamboo soil bacterial community was the most diverse, followed by the transition zone, with the cedar plantation possessing the lowest diversity. The results from both techniques revealed that the Acidobacteria and Proteobacteria predominated in the three communities, though the relative abundance was different. The 250 most abundant OTUs represented about 70 % of the total sequences found by pyrosequencing. Most of these OTUs were found in all three soil communities, demonstrating the overall similarity among the bacterial communities. Nonmetric multidimensional scaling analysis showed further that the bamboo and transition soil communities were more similar with each other than the cedar soils. These results suggest that bamboo invasion to the adjacent cedar plantation gradually increased the bacterial diversity and changed the soil community. In addition, while the 10 most abundant OTUs were distributed worldwide, related sequences were not abundant in soils from outside the forest studied here. This result may be an indication of the uniqueness of this region.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soil microbial communities play an important role in nutrient cycling and organic matter turnover and are essential components in forest ecosystems [1, 2]. Different forest types as well as environmental factors can alter the structure of the soil microbial community. Native and plantation forest soils can possess distinct microbial communities [1, 3]. Changing dominant tree species also clearly influences the decomposer community composition [4]. These influences may be attributed to tree species differences in litter quality, root exudates, and nutrient availability [5–7]. Soil microorganisms in turn can affect plant development, plant community composition, and ecosystem function [8, 9].
Bamboos are one of the most important forest resources in East Asia. They are commonly planted to produce timber for construction, furniture, and other industries. Young bamboo shoots are also in high demand as a healthy food with a delicious taste and high fiber content. Bamboos grow by producing below-ground rhizomes that spread laterally and generate bamboo sprouts at the soil surface. Hence, bamboo colonies rapidly expand. Common management practices to maintain and increase bamboo production include regular removal of understory vegetation, tillage, and fertilizer application. These practices may increase soil CO2 efflux [10], increase the water-soluble organic N concentration in soils [11], and reduce soil microbial functional diversity [12].
Expansion of bamboo forests in broad-leaved and/or coniferous forests can reduce tree diversity and alter the vegetation type. With the rhizome system and fast-growing characteristics, bamboo can invade adjacent forests. High densities of bamboo stalks cause shading and retard the regeneration for other young trees. Gradually, the bamboo dominates the expanded territory. Several studies have focused on the ecological effects of bamboo invasions, including the mechanism of its invasion into secondary broad-leaved forest [13], impacts on surface runoff [14], changes in dissolved soil organic carbon and nitrogen contents [11], and changes in the myrmecofauna [15]. Bamboos can release allelochemicals from their leaves [16]. Their allelopathy reduces seedling abundance and species richness under bamboo canopies and in turn causes changes in plant community composition and species diversity [17]. However, there are few reports on the impact of bamboo invasion on the structure and diversity of the bacterial community of forest soils. Therefore, the objectives of this study were to investigate the soil bacterial community of bamboo forest and evaluate the effects of bamboo invasion in an adjacent coniferous plantation on soil bacterial community structure. The indigenous soil bacterial communities were phylogenetically characterized by using a combination of 16S rRNA gene clone libraries and bar-coded pyrosequencing technique analysis. Thus, this study provides insights into the structure and diversity of bamboo soil bacterial community and information on the biogeochemical processes in this invasive ecosystem.
Materials and Methods
Site Description and Soil Sampling
This study was conducted at Shanlinshi, a subtropical montane area in Nantou County, central Taiwan (23°40′ N, 120°46′ E). The elevation is about 1,350 m a.s.l., the mean annual precipitation is about 2,600 mm, and the mean annual temperature is 17 °C. Parts of this area were reforested with Japanese cedar (Cryptomeria japonica) (Supplementary Fig. 1a) about 40 years ago after large-scale cutting of the natural camphor forest. Almost at the same time, Moso bamboo (Phyllostachys edulis), a temperate species of giant timber bamboo, was introduced and established adjacent to the cedar plantation (Supplementary Fig. 1a). Currently, a transition zone, with both cedar and bamboo plants of 30 to 50 m wide, stretches in the boundary between the bamboo and cedar plantations (Supplementary Fig. 1b). Farmers occasionally cut bamboo stems to induce the regeneration of bamboo, leaving many stumps in the bamboo forest (Supplementary Fig. 1c). In contrast, the soil in cedar plantation is largely undisturbed and has lush understory plants (Supplementary Fig. 1d).
Three parallel transect lines separated by more than 50 m at each forest type were surveyed from moso bamboo forest, the transition zone, and cedar plantation in February 2011. Although the study site is located in humid mountain cloud region, the high evaporation sometimes causes drought in the summer. In order to avoid the fortuitous changes of microbial communities caused by fluctuation of soil moisture, we collected soil samples in winter. The soil samples were collected in the same time to avoid difference caused by seasonal change. At each vegetation type of the transect lines, three 25 m × 25 m plots were set up. Within each plot, three subsamples were collected with a soil auger 8 cm in diameter and 10 cm deep and were combined. Visible detritus materials, such as roots and litter, were manually removed prior to passing soil through a 2-mm sieve. Soils were then stored at −20 °C for a few days before DNA extraction.
The soils were characterized as Hapalorthepts, which were clayey loam and usually moderately well drained. The parent material is sandstone. The pH and other properties are reported in Table 1.
16S rRNA Gene Clone Library Construction and Sequencing
The 16S rRNA clone libraries for each of the nine samples were constructed as described previously in Lin et al. [18]. In brief, soil community DNA was extracted using the PowerSoil® Soil DNA Isolation kit (MoBio Industries, Carlsbad, CA, USA) in accordance with manufacturer's instructions. The bacterial 16S rRNA genes were amplified by polymerase chain reactions (PCR) with the primer set 27 F and 1492R [19]. After 15 cycles, the PCR products were cloned using the TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA) and the pCR2.1 vector. White colonies on selective Luria–Bertani (LB) agar plates were picked into 96-well blocks containing 1 ml of LB broth plus kanamycin (50 μg ml−1) and grown overnight. Sterile glycerol was added to a final concentration of 10 %, and an aliquot of this was transferred to a 96-well sequencing block. Both the sequencing and the original culture blocks were stored at −80 °C. Bacterial clones were partially sequenced using primer 27 F. Sequence analysis was performed using an ABI PRISM Big Dye Terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA) and an ABI 3730 Genetic Analyzer (Applied Biosystems) following the manufacturer's instructions. Sequences were analyzed with the Mallard and Pintail programs to test for chimeras [20, 21]. The entire clone sequences obtained in the present study have been deposited into GenBank database under accession numbers JN851193-JN851701.
Bar-Coded Pyrosequencing of the 16S rRNA Genes
The V1 to V2 regions of the bacterial 16S rRNA gene were amplified using 27 F and 338R primers [19]. The targeted region has been shown to be appropriate for the accurate taxonomic classification of bacterial sequences [22]. PCR were performed as following: 94 °C for 3 min followed by 20 cycles of 94 °C for 45 s; 50 °C for 30 s and 72 °C for 90 s; and a final elongation at 72 °C for 10 min. Secondary PCR (3 cycles rather than 20) was carried out using primers with different bar code sequences as: adaptor A-linker-27 F and adaptor B-linker-8-bp bar code-338R. The bar code for each sample was unique and error-correcting to facilitate sorting of sequences from a single pyrosequencing run [23]. The bar-coded PCR products were then purified using the PCR cleanup system (Viogene Biotek Corp., New Taipei City, Taiwan). The qualities and concentrations of the purified bar-coded PCR products were determined using a NanoDrop Spectrophotometer (Thermo Scientific). Amplicon pyrosequencing was performed by Mission Biotech (Taipei, Taiwan) using the 454/Roche GS-FLX Titanium Instrument (Roche, NJ, USA). All sequences have been submitted to the Short Read Archives (SRA) with the accession number of SRA064346.
Sequence Analysis
Taxonomic assignment of sequences from the clone library was made using the naïve Bayesian rRNA classifier [24] in Ribosomal Database Project (RDP) (http://rdp.cme.msu.edu/index.jsp). Diversity estimates, including evenness, Shannon diversity index, Chao1 estimator, and rarefaction analyses, were calculated by the program DOTUR [25]. The pyrosequences were processed through the RDP pyrosequencing pipeline (http://pyro.cme.msu.edu). The total sequences were assigned to each sample by recognition of the bar code, and those shorter than 200 bp or low quality were deleted. Taxonomic information was analyzed using the naïve Bayesian rRNA classifier [24] of the RDP. Rarefaction curves were calculated by using the programs Aligner, Complete Linkage Clustering, and Rarefaction of the RDP pyrosequencing pipeline. Diversity indices, including Shannon diversity index and Chao1 estimator, were calculated based on the Complete Linkage Clustering data for operational taxonomic units (OTUs) with an evolutionary distance of 0.03. Venn diagrams of shared OTUs among the three communities were obtained using the Mothur program [26]. The PRIMER V6 software (PRIMER-E, Lutton, Ivybridge, UK) was used for nonmetric multidimensional scaling (NMDS) generated with Bray–Curtis similarity of pyrosequencing phylotype data.
Results
Bacterial Community Analyzed Using Clone Library
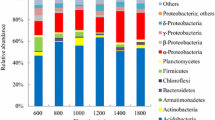
About 50 to 60 clones of 16S rRNA genes were derived from each replicate sample. The sequences from three replicate samples in each study site were then combined for further analysis. A total of 169 bacterial sequences were obtained for bamboo, 177 for transition zone, and 166 for cedar plantation. Three chimeric sequences from transition soils were detected, and the remaining sequences were used for further analyses. Phylogenetic analysis revealed that the three communities were composed of eight bacterial groups. The Acidobacteria- (comprising 63, 58, and 56 % of all clones from bamboo, transition, and cedar, respectively) and Proteobacteria-affiliated clones (25, 39, and 27 %, respectively) were the two most abundant phyla (Fig. 1a). Minor phyla included Actinobacteria, Bacteroidetes, Firmicutes, Planctomycetes, and Verrucomicrobia, which were all less than 7.2 % of the clones (Fig. 1a). Within the Proteobacteria, α-Proteobacteria was the most abundant group and included mostly Rhizobiales in all three communities.

Relative abundance of phylogenetic groups analyzed using a clone library and b pyrosequencing methods in three forest soil communities
For calculation of diversity indices, OTUs were formed at an evolutionary distance ≤0.03 (about 97 % sequence similarity). Based on the number of OTUs, Shannon diversity index and Chao1 estimator, the soil bacterial community of the bamboo soils was more diverse than the other two communities (Table 1). The rarefaction curve analysis also revealed similar results, where the slope of curve of the bamboo soil community was steeper than that in the cedar soils, and the transition community was intermediate (Fig. 2a).

Rarefaction curve analysis using a clone library and b pyrosequencing methods for three forest soil communities. OTUs were defined as sequences sharing 97 % nucleotide sequence similarity
Bacterial Community Analyzed Using Pyrosequencing
To explore the phylogenetic structure and diversity of soil bacterial community with increased resolution, bar-coded pyrosequencing of the 16S rRNA genes present in the community DNAs was performed. About 3,000–7,000 sequences were obtained from each replicate. A total of 12,465, 13,016, and 11,964 sequences were obtained from bamboo, transition, and cedar forest soils, respectively. These sequences were classified into 20 phylogenetic groups. The phyla Acidobacteria (47–53 %) and Proteobacteria (28–34 %) also predominated among the three communities (Fig. 1b). Within the Proteobacteria, α-Proteobacteria was also the most abundant group and mainly associated with Rhizobiales and Rhodospirillales in all three communities. The proportions of other members, such as Actinobacteria, Bacteroidetes, Chloroflexi, and Firmicutes, were relatively low (<7 %) (Fig. 1b).
Using the pyrosequencing method, 2,268, 2,078, and 1,632 OTUs, based on the cutoff value of 0.03 for the genetic distance, were identified in the bamboo, transition, and cedar soil communities, respectively. Besides the number of OTUs, Shannon diversity index, Chao1 estimator, and rarefaction curve analysis also indicated that bamboo soil was more species rich than the soils of the other two communities (Table 2; Fig. 2). Though a high number of sequences were obtained, the rarefaction curves indicated that the community diversity did not reach an asymptote in the present study. Soils from the bamboo forest had the highest diversity, followed by soils from the transition and cedar forest.
Although the overall diversity observed was much greater than observed in the clone libraries, the relative diversity of the three soil communities was similar.
Comparison of the Bacterial Communities Observed by the Clone Library and Pyrosequencing Analyses
About 85 % of the OTUs in the clone libraries were also present in the pyrosequencing data sets. Given the vastly larger size of the pyrosequencing data sets, the OTUs only found in the clone libraries indicated that one or both of the methods was biased, presumably by the differences in the primers utilized between two techniques. On the other hand, given the similarities in the overall composition and diversity of the bacterial communities observed, these biases did not obscure many properties of the communities. In contrast, as expected from its much larger size, more than 97 % of the OTUs in the pyrosequencing data sets were absent from the clone libraries, indicating the advantages of pyrosequencing for deep surveys of soil microbial communities.
The compositions of both the clone and pyrosequencing libraries were similar. Both methods indicated that the Acidobacteria was the most abundant phylum in all three communities and represented more than half of the communities. Similarly, within the Proteobacteria, the α-Proteobacteria predominated in all three communities analyzed using either method. However, in the clone libraries, the abundance of the γ-Proteobacteria sequences was much higher in the transition community and lower in the cedar soils than found using pyrosequencing. In contrast, 5–7 % of the pyrosequencing sequences grouped within the unclassified Proteobacteria, which included sequences that did not cluster into any of the recognized proteobacterial divisions. This category was not observed in the clone libraries. Similarly, the phylum Actinobacteria represented 6 % of the pyrosequencing sequences and less than 1 % in the clone libraries. Lastly, the phyla Chloroflexi, Cyanobacteria, Gemmatimonadetes, Lentisphaerae, and Nitrospira were only retrieved by using the pyrosequencing method. Because of their low relative abundance, they would not be expected to be found in the clone libraries.
Bacterial Community Comparison
Although there were differences in community diversity, the compositions of the bacterial communities of the three forest soils were similar, especially that between bamboo and transition soils. The 250 most abundant OTUs represented about 70 % of the total sequences found by pyrosequencing. Most of these OTUs were found in all three soil communities, demonstrating the overall similarity among the bacterial communities (Table 3 and additional data shown in Supplementary Table 1). Nonmetric multidimensional scaling analysis showed further that the bamboo and transition soil communities were more similar with each other than the cedar soils (Fig. 3). This analysis was supported by the observation that some of the abundant OTUs, such as numbers 4, 168, 17, 63, 119, 48, 379, 124, and 489, were several times higher in both the bamboo and transition soils than in the cedar community. Likewise, OTUs 33, 115, 45, 126, 179, 166, 168, 142, 108, and 105 were more abundant in the cedar community than the other soils. Thus, the differences in composition were a matter of relative abundance and not present and absence of specific bacterial groups.

NMDS plot of three forest soil bacterial communities based on the 250 most abundant OTUs (about 70 % of all sequences) of the pyrosequencing data. The OTUs were formed at an evolutionary distance of 0.03
Within the pyrosequencing data, the 10 most abundant OTUs comprised about 20 % of all sequences and were clustered into Acidobacteria, Actinobacteria, and α- and γ-Proteobacteria (Table 3). Each of them comprised about 1–5 % of the community. Most of these abundant OTUs were also present in the clone libraries. Among these 10 OTUs, numbers 81, 12, 33, 127, 69, and 27 were closely related (99–100 % similarity) to sequences retrieved from different ecosystems worldwide (Table 3). However, most of these related sequences were not among the most abundant in these other ecosystems. In contrast, two OTUs were not widely distributed. An acidobacterial-affiliated OTU, no. 145, was only closely related to clones from this study site. It was more abundant in the bamboo and transition soils. The similarities with sequences from other study sites were all less than 97 %. The most abundant OTU at this site, no. 4, was also only related to sequences from other study sites with similarity of less than 98 %.
Discussion
Our results reveal that the bamboo invasion mainly increases the bacterial diversity of the bamboo soil community. Acidobacteria and Proteobacteria were the two most abundant phyla in the three communities, although their relative abundances were different. The distribution of some abundant OTUs was also different. It is known that different tree species, e.g., broad-leaved versus conifer trees, have different leaf and litter chemistry. They can alter not only soil physicochemical properties but also microbial community and enzyme activities [27, 28]. Thus, the soil properties, such as C and N content, in transition zone were more similar to those in bamboo plantation (Table 1). In addition, the cedar plantation has lush understory plants, which could result higher soil C and N content [29]. Understory plants and their rhizosphere resources, including root exudates and nutrients, also influence the structure of variation in the soil microbial community [30]. The collection of bamboo shoots by farmers causes disturbance of soil. This process is like tillage which might reduce the content of soil organic matter and subsequently change the bacterial communities [31, 32]. By comparison, the soil in cedar plantation was largely undisturbed. Soils in cedar plantation were more acidic than other two forests. Soil pH could be another factor to shape the difference of soil bacterial communities between bamboo forest and cedar plantation [33]. In addition, using phospholipid fatty acid analysis, we also found significant separation of fungal communities between bamboo and cedar soils (unpublished data). Other study also revealed that vegetation type change altered soil fungal community [34].
Allelopathic effects could be another important factor structuring the soil communities. Tsai and Young [35] identified 11 phenolic compounds from the soil beneath bamboo Denrocalamus latiflorus Munro. In moso bamboo, six allelochemicals were found in the aqueous leachate and extracts of leaves and alcoholic soil extracts [16]. These allelochemicals could also alter the soil microbial community as well as the growth of the neighboring plants. Using a diffusion method, Rauha et al. [36] showed that some plant phenolics were effective against bacterial growth, and the phyllosphere bacterial abundance was negatively correlated with total phenolics content of the leaf, mesophyll, and abaxial epidermis [37]. Similarly, using denaturing gradient gel electrophoresis analysis, the bacterial community of a broad-leaved forest soil differed from that following a bamboo invasion [38]. On the other hand, the allelopathic compounds from invasive plant leachates lead to significant change in soil functional diversity but did not affect the bacterial structure in pine forest. The allelopathic effects could depend on the ecosystems studied [39]. As small change in community composition between bamboo and cedar forest soils, further study in microbial functional profiles is needed to verify the changes in the bamboo-invaded ecosystem.
In the three sites, the diversity of soil bacterial community of moso bamboo forests was higher than that in the other two soil communities, similar to the previous results [40]. For harvesting timber and young bamboo shoots, management practices in moso bamboo forest disturb the surface soils, and disturbances of forest soils increase the diversity of their microbial communities [3, 40, 41]. Bacterial communities in frequently tilled cultural soils are also more diverse than in less disturbed forest soils [31, 32]. Following disturbances, surface soils might be subjected to fluctuations of soil moisture and temperature, which may also increase the diversity of bacterial community. Disturbances could also increase the loss of organic matter and result in the low content of organic C and total N in bamboo soils. Low organic matter in soils is also correlated with a decline in diversity in many soil bacterial communities [42]. Presumably, disturbance is a stronger influence in these soils at this time.
Using pyrosequencing technique, some of the 10 most abundant OTUs among the three communities were distributed widely in different ecosystems, including those from temperate, subtropical, and tropical forests, cropping system at Taiwan, Europe [43, 44], North and South America [45], even Antarctica [46], though their relative abundance was low. The results indicate that these OTUs are distributed worldwide. Two of the 10 most abundant OTUs, in contrast, are not widely distributed. Compared to the OTUs mentioned above, the absence of these two OTUs from other data sets is intriguing. Although methodological biases may have excluded these OTUs in other studies and sampling of soils worldwide is still incomplete, it also possible that they are endemic to Taiwan or abundant largely in bamboo and bamboo-invaded forest soil communities or subtropical montane forests. In addition, most of these 10 abundant OTUs were also present in the clone libraries from the same soils, indicating that their abundance is not method dependent and the primer set is suitable for soil community surveys.
The phylum Acidobacteria comprised more than half of the sequences from bamboo soils using both clone library and pyrosequencing analysis. Other studies have also found a wide distribution among various soil communities, including that in agricultural system, subtropical, and tropical forests [31, 47, 48]. It is generally believed that Acidobacteria are oligotrophs [49] and versatile heterotrophs, exhibiting slow metabolic rates under low-nutrient conditions [50]. Using culture-dependent methods, no Acidobacteria-affiliated isolates could be retrieved from bamboo soils [51]. It is worthwhile to further explore their function in the bamboo and bamboo-invaded soil communities.
Proteobacteria were also abundant in all three soil communities, as well as those in other soils [52]. In the three communities, α-Proteobacteria is the most abundant class of Proteobacteria, and many sequences are related to the orders Rhizobiales and Rhodospirillales. They include species attributed to nitrogen fixation, organic matter decomposition, and plant growth promotion [53, 54]. In the β-Proteobacteria, Burkholderia is also common in all three communities. This genus includes species capable of fixing nitrogen, promoting plant growth, and acting as biocontrol agents [55]. The physiological properties of these microbes might affect soil function in different ecosystems.
In this study, Pseudomonas-related sequences were rare and only found with very low abundance in the bamboo forest soils. In other studies of bamboo forest soils, no members of this genus were isolated from the rhizosphere [51, 56]. The pseudomonads are important components of the root-associated microbial community. They can prevent infection by plant pathogens and improve plant growth [57, 58]. They also degrade phenolic allelochemicals [59]. It is not known why the abundance of this genus is so low in the three communities and the influence of this phenomenon on the metabolic ability of the bamboo or cedar soil bacterial communities. Further study of their functional diversity and isolation of more representatives will be worthwhile to address these questions of the microbial community in bamboo soils.
The relative abundance of Bacteroidetes in the cedar community is higher than that in bamboo and transition soils. Known as copiotrophs, Bacteroidetes tend to be found in nutrient-rich environments and have the ability to degrade organic matter [60]. Considering the high amount of organic carbon in cedar soils, it could provide a favorable condition for the growth of Bacteroidetes. In comparison, the growth of Bacteroidetes may be limited by the lower levels of organic matter content in the frequently disturbed bamboo soils. Moreover, Hortal et al. [29] reported that presence of shrubs were associated with the relative abundance of Bacteroidetes. Thus, the lush understory plants in the cedar plantation may be correlated with the higher abundance of Bacteroidetes than that in other two communities.
In conclusion, conversion of a cedar to a bamboo plantation correlated with changes in the diversity and composition of the soil bacterial community. The increase in diversity was consistent with increased soil disturbances resulting from the more intensive management of the bamboo plantation. The changes in composition of the bacterial communities were small, possibly resulting from either direct or indirect effects of the allelochemicals produced by moso bamboo. The composition of the bamboo-invaded transition community was more similar to the bamboo soils than to the cedar soils. Although the pyrosequencing technique provides a new powerful tool to investigate the bacterial community of bamboo forest soils, further comparisons of differences in functional diversity between bamboo and adjacent forest of soil biota are also needed to fully elucidate the ecological effects of bamboo invasion to forest ecosystem.
References
Burton J, Chen C, Xu Z, Ghadiri H (2010) Soil microbial biomass, activity and community composition in adjacent native and plantation forests of subtropical Australia. J Soils Sediments 10:1267–1277
Doran JW, Zeiss MR (2000) Soil health and sustainability: managing the biotic component of soil quality. Appl Soil Ecol 15:3–11
Lin YT, Jangid K, Whitman WB, Coleman DC, Chiu CY (2011) Change in bacterial community structure in response to disturbance of natural hardwood and secondary coniferous forest soils in central Taiwan. Microb Ecol 61:429–437
Kubartová A, Moukoumi J, Béguiristain T, Ranger J, Berthelin J (2007) Microbial diversity during cellulose decomposition in different forest stands: I. Microbial communities and environmental conditions. Microb Ecol 54:393–405
Grayston SJ, Prescott CE (2005) Microbial communities in forest floors under four tree species in coastal British Columbia. Soil Biol Biochem 37:1157–1167
Quideau SA, Chadwick OA, Benesi A, Graham RC, Anderson MA (2001) A direct link between forest vegetation type and soil organic matter composition. Geoderma 104:41–60
Xu Z, Ward S, Chen C, Blumfield T, Prasolova N, Liu J (2008) Soil carbon and nutrient pools, microbial properties and gross nitrogen transformations in adjacent natural forest and hoop pine plantations of subtropical Australia. J Soils Sediments 8:99–105
Batten K, Scow K, Espeland E (2008) Soil microbial community associated with an invasive grass differentially impacts native plant performance. Microb Ecol 55:220–228
Merilä P, Malmivaara-Lämsä M, Spetz P, Stark S, Vierikko K, Derome J, Fritze H (2010) Soil organic matter quality as a link between microbial community structure and vegetation composition along a successional gradient in a boreal forest. Appl Soil Ecol 46:259–267
Liu J, Jiang P, Wang H, Zhou G, Wu J, Yang F, Qian X (2011) Seasonal soil CO2 efflux dynamics after land use change from a natural forest to moso bamboo plantations in subtropical China. Forest Ecol Manag 262:1131–1137
Wu JS, Jiang PK, Chang SX, Xu QF, Lin Y (2010) Dissolved soil organic carbon and nitrogen were affected by conversion of native forests to plantations in subtropical China. Can J Soil Sci 90:27–36
Xu Q, Jiang P, Xu Z (2008) Soil microbial functional diversity under intensively managed bamboo plantations in southern China. J Soils Sediments 8:177–183
Okutomi K, Shinoda S, Fukuda H (1996) Causal analysis of the invasion of broad-leaved forest by bamboo in Japan. J Veg Sci 7:723–728
Ide J, Shinohara Y, Higashi N, Komatsu H, Kuramoto K, Otsuki K (2010) A preliminary investigation of surface runoff and soil properties in a moso-bamboo (Phyllostachys pubescens) forest in western Japan. Hydro Res Lett 4:80–84
Touyama Y, Yamamoto T, Nakagoshi N (1998) Myrmecofaunal change with bamboo invasion into broadleaf forests. J For Res 3:155–159
Chou CH, Yang CM (1982) Allelopathic research of subtropical vegetation in Taiwan II. Comparative exclusion of understory by Phyllostachys edulis and Cryptomeria japonica. J Chem Ecol 8:1489–1507
Larpkern P, Moe SR, Ørjan T (2011) Bamboo dominance reduces tree regeneration in a disturbed tropical forest. Oecologia 165:161–168
Lin YT, Huang YJ, Tang SL, Whitman WB, Coleman DC, Chiu CY (2010) Bacterial community diversity in undisturbed perhumid montane forest soils in Taiwan. Microb Ecol 59:369–378
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 115–175
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2005) At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71:7724–7736
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2006) New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72:5734–5741
Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. Nucl Acids Res 35:e120
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5:235–237
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian Classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartman M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Lejon D, Chaussod R, Ranger J, Ranjard L (2005) Microbial community structure and density under different tree species in an acid forest soil (Morvan, France). Microb Ecol 50:614–625
Ushio M, Kitayama K, Balser TC (2010) Tree species-mediated spatial patchiness of the composition of microbial community and physicochemical properties in the topsoils of a tropical montane forest. Soil Biol Biochem 42:1588–1595
Hortal S, Bastida F, Armas C, Lozano YM, Moreno JL, García C, Pugnaire FI (2013) Soil microbial community under a nurse-plant species changes in composition, biomass and activity as the nurse grows. Soil Biol Biochem 64:139–146
McIntosh ACS, Macdonald SE, Quideau SA (2013) Linkages between the forest floor microbial community and resource heterogeneity within mature lodgepole pine forests. Soil Biol Biochem 63:61–72
Jangid K, Williams MA, Franzluebbers AJ, Sanderlin JS, Reeves JH, Jenkins MB, Endale DM, Coleman DC, Whitman WB (2008) Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol Biochem 40:2843–2853
Upchurch R, Chiu CY, Everett K, Dyszynski G, Coleman DC, Whitman WB (2008) Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol Biochem 40:1294–1305
Rousk J, Bååth E, Brookes PC, Lauber C, Lozupone C, Caporaso JG, Knight R, Fierer N (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4:1340–1351
Curlevski NJA, Xu Z, Anderson IC, Cairney JWG (2010) Converting Australian tropical rainforest to native Araucariaceae plantations alters soil fungal communities. Soil Biol Biochem 42:14–20
Tsai CS, Young CC (1993) Allelochemicals in rhizosphere soils of flowering and nonflowering bamboo plants. Bot Bull Acad Sin 34:223–234
Rauha JP, Remes S, Heinonen M, Hopia A, Kähkönen M, Kujala T, Pihlaja K, Vuorela H, Vuorela P (2000) Antimicrobial effects of Finnish plant extracts containing flavonoids and other phenolic compounds. Int J Food Microbiol 56:3–12
Yadav R, Karamanoli K, Vokou D (2005) Bacterial colonization of the phyllosphere of Mediterranean perennial species as influenced by leaf structural and chemical features. Microb Ecol 50:185–196
Wang Q, Xu Q, Jiang P, Qin H (2009) DGGE analysis of PCR of 16S rDNA V3 fragment of soil bacteria community in soil under natural broadleaf forest invaded by Phyllostachys pubescens in Tianmu Mountain National Nature Reserve. Acta Pedo Sin 46:662–669
Lorenzo P, Pereira CS, Rodrguez-Echeverra S (2013) Differential impact on soil microbes of allelopathic compounds released by the invasive Acacia dealbata Link. Soil Bio Biochem 57:156–163
Jangid K, Williams MA, Franzluebbers AJ, Blair JM, Coleman DC, Whitman WB (2010) Development of soil microbial communities during tallgrass prairie restoration. Soil Biol Biochem 42:302–312
Lin YT, Jangid K, Whitman WB, Coleman DC, Chiu CY (2011) Soil bacterial communities in native and regenerated perhumid montane forests. Appl Soil Ecol 47:111–118
Tripathi BM, Kim M, Singh D, Lee-Cruz L, Lai-Hoe A, Ainuddin AN, Go R, Rahim RA, Husni MHA, Chun J, Adams JM (2012) Tropical soil bacterial communities in Malaysia: pH dominates in the equatorial tropics too. Microb Ecol 64:474–484
Stephanie AE, Kuske CR, Schmidt TM (2011) Influence of plant polymers on the distribution and cultivation of bacteria in the phylum Acidobacteria. Appl Environ Microbiol 77:586–596
Hui N, Liu XX, Kurola J, Mikola J, Romantschuk M (2012) Lead (Pb) contamination alters richness and diversity of the fungal, but not the bacterial community in pine forest soil. Boreal Environ Res 17:46–58
Dunber J, Eichorst SA, Gallegos-Graves LV, Silva S, Xie G, Hengartner NW, Evans RD, Hungate BA, Jackson RB, Megonigal JP, Schadt CW, Vilgalys R, Zak DR, Kuske CR (2012) Common bacterial responses in six ecosystems exposed to 10 years of elevated atmospheric carbon dioxide. Environ Microbiol 14:1145–1158
Yergeau E, Bokhorst S, Kang S, Zhou J, Greer CW, Aerts R, Kowalchuk GA (2012) Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J 6:692–702
Araujo JF, de Castro AP, Costa MMC, Togawoa RC, Pappas Júnior GJ, Quirino BF, Bustamante MMC, Williamson L, Handelsman J, Krüger RH (2012) Characterization of soil bacterial assemblies in Brazilian savanna-like vegetation reveals Acidobacteria dominance. Microb Ecol 64:760–770
Meng H, Li K, Nie M, Wan JR, Quan ZX, Fang CM, Chen JK, Gu JD, Li B (2013) Responses of bacterial and fungal communities to an elevation gradient in a subtropical montane forest of China. Appl Microbiol Biotechnol 97:2219–2230
Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL, Townsend AR (2010) Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem 42:2153–2160
Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM et al (2009) Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75:2046–2056
Han J, Xia D, Li L, Sun L, Yang K, Zhang L (2009) Diversity of culturable bacteria isolated from root domains of moso bamboo (Phyllostachys edulis). Microb Ecol 58:363–373
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728
Zhang L, Xu Z (2008) Assessing bacterial diversity in soil. J Soils Sediments 8:379–388
Yarwood SA, Myrold DD, Högberg MN (2009) Termination of below-ground C allocation by tree alters soil fungal and bacterial communities in a boreal forest. FEMS Microbiol Ecol 70:151–162
Coenye T, Vandamme P (2003) Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ Microbiol 5:719–729
Li L, Liu M, Yang S, Liu L, Miao K, Yang K, Han J (2008) Cultivable microbial diversity at the rhizosphere of Phyllostachys pubescens. Acta Microbiol Sin 48:772–779
Fett WF (2006) Inhibition of Salmonella enterica by plant associated pseudomonads in vitro and on sprouting alfalfa seed. J Food Prot 69:719–728
Rangarajan S, Loganathan P, Saleena LM, Nair S (2001) Diversity of pseudomonads isolated from three different plant rhizospheres. J Appl Microbiol 91:742–749
Zhang ZY, Pan LP, Li HH (2010) Isolation, identification and characterization of soil microbes which degrade phenolic allelochemicals. J Appl Microbiol 108:1839–1849
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Acknowledgments
This work was supported by the Taiwan National Science Council, Taiwan (NSC 101-2621-B-001-002-MY3).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(XLSX 31 kb)
Supplementary Fig. 1
(a) Aerial view of cedar (dark green) and bamboo forests (light green). (b) Bamboo-invaded transition zone. (c) Bamboo forest. Bamboo stems were occasionally cut by farmers and lot of stumps are in the forest. (d) Cedar forest. This forest is largely undisturbed and has lush understory plants. (e) Cross view from cedar (left) to bamboo (right) forest (DOC 771 kb)
Rights and permissions
About this article
Cite this article
Lin, YT., Tang, SL., Pai, CW. et al. Changes in the Soil Bacterial Communities in a Cedar Plantation Invaded by Moso Bamboo. Microb Ecol 67, 421–429 (2014). https://doi.org/10.1007/s00248-013-0291-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-013-0291-3