
Abstract
Community level physiological profiling and pyrosequencing-based analysis of the V1-V2 16S rRNA gene region were used to characterize and compare microbial community structure, diversity, and bacterial phylogeny from soils of chemically cultivated land (CCL), organically cultivated land (OCL), and fallow grass land (FGL) for 16 years and were under three different land use types. The entire dataset comprised of 16,608 good-quality sequences (CCL, 6,379; OCL, 4,835; FGL, 5,394); among them 12,606 sequences could be classified in 15 known phylum. The most abundant phylum were Proteobacteria (29.8%), Acidobacteria (22.6%), Actinobacteria (11.1%), and Bacteroidetes (4.7%), while 24.3% of the sequences were from bacterial domain but could not be further classified to any known phylum. Proteobacteria, Bacteroidetes, and Gemmatimonadetes were found to be significantly abundant in OCL soil. On the contrary, Actinobacteria and Acidobacteria were significantly abundant in CCL and FGL, respectively. Our findings supported the view that organic compost amendment (OCL) activates diverse group of microorganisms as compared with conventionally used synthetic chemical fertilizers. Functional diversity and evenness based on carbon source utilization pattern was significantly higher in OCL as compared to CCL and FGL, suggesting an improvement in soil quality. This abundance of microbes possibly leads to the enhanced level of soil organic carbon, soil organic nitrogen, and microbial biomass in OCL and FGL soils as collated with CCL. This work increases our current understanding on the effect of long-term organic and chemical amendment applications on abundance, diversity, and composition of bacterial community inhabiting the soil for the prospects of agricultural yield and quantity of soil.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soil microorganisms are highly diverse and abundant organisms on earth; 1 g of soil may contain billions of microbes with thousand of different species [48]. These microbes play a pivotal role in the decomposition of plant and animal organic matter for plant growth and soil structure and fertility. Several biotic or abiotic factors lead to the alteration of microbial community structure and composition which may directly or indirectly influence the soil ecosystem, nutrient cycle activity, and crop production [7, 32, 43, 52]. In addition, anthropogenic intervention for the management and treatment of soil via pesticide [17], chitin [16], compost, manure [14, 41], or genetically modified microorganism and plant [5] also influence microbial diversity. Thus maintenance of microbial diversity and composition is very important for the sustainable agricultural production. Soils under organic farming (compost and green manure) have better quality and microbial activity [32] due to crop rotation and reduced application of synthetic nutrients and pesticides in organically managed soil [42, 51]. Chemical fertilizers (nitrogen, phosphorus, and potassium) enhance crop yield but also bring alteration in soil properties, functional diversity in microbial population, and their enzymatic activities [19, 20]. The long-term experiments demonstrated that initial restoration successes may be transient, so monitoring after long term is crucial for restoration experiments, especially when results can be influenced strongly by time effects [24]. Several studies have been made to evaluate how the application of fresh and composted organic wastes modifies the structure, size, and activity of soil microbial community [2]. Although long-term impact of chemical fertilizers on soil microbial biomass and diversity is not well documented, it has been shown that chemical fertilizers could increase the soil microbial biomass C and N [15, 45], as well as no significant change in the microbial characteristics of the soil [24]. Zhong and Cai [53] suggested that changes in microbial parameters are correlated with the soil organic carbon content and not to the application of P and N. Evidences linking direct impact of chemical fertilizers on microbial diversity function and phylogeny are not so evident [53].
In the current study, we have compared microbial community structure, diversity, and bacterial phylogeny in chemically cultivated land (CCL), organically cultivated land (OCL), and an adjacent land left as fallow grassland (FGL), where no cultivation have been practiced in last 16 years. OCL under organic farming was fortified annually with composted organic manure (primarily of cow dung) and no tillage and no crop residues were removed for the same period of time, whereas in CCL, chemical fertilizers in form of nitrogen, phosphorus, and potassium were applied. State of the art technology, community level physiological profiling (CLPP), and recently developed 16S rRNA gene-based high throughput sequencing (454 pyrosequencing) were employed to understand the functional microbial diversity and underlying phylogenetic changes in response to long-term use of chemical and organic fertilizers into the farm soil.
Materials and Methods
Site Description and Soil Sampling
Soil samples were collected from three different fields: (1) treated with chemical fertilizers (CCL); (2) OCL; and (3) untreated for past 16 years (FGL), of Dorli village of Yavatmal District (19.26′ and 20.42′ N, 77.18′ and 79.9′ E) of Maharashtra State of India. Maximum and minimum temperature ranges between 45.6°C and 5.6°C, with mean annual rainfall of 10,566 mm. Due to soil degradation processes such as wind and water erosion, organic matter decline, soil compaction, and soil fertility loss occur rapidly in these areas because of high temperatures and low and irregular precipitation. The region is prone to recurring droughts. Crops grown in the Yavatmal District depend on natural precipitation and therefore, mainly grain legumes are cultivated.
OCL received about 20 tons of composted cow manure per hectare. Thus natural grassland was converted to cropland growing grain legume crops under organic management. No tillage and crop residue removal been practiced since the past 16 years. Adjacent land of 1- ha size was left uncultivated for permanent FGL, as described earlier [32]. In CCL, chemical fertilizers were applied in form of elements of nitrogen, phosphorus, and potassium in 60:30:30 kg/ha ratio, respectively. The fertilizers were applied annually for the past 16 years. Within each from CCL, FGL, and OCL, four 10 × 10-m areas were selected for uniform topology and soil type. Soil samples were collected randomly from three places, at the depth of 15 cm, in each of the four selected areas in October 2010. Three samples collected from four areas were randomly mixed, and the composite was considered as the three replicates for analysis. Soil samples were kept in plastic bags on ice and transported to the laboratory and stored at 4°C. All the biological assays were conducted within 48 h. Soil samples were sieved to pass 2-mm round-holed sieve. Visible organic matter was removed before analysis, and the residual soil was air-dried for chemical analysis.
Soil Analysis
Physical and chemical properties of soil were determined as described by Marinari et al. [29]. Organic carbon was determined by dichromate oxidation [33]. Total available nitrogen was determined by Kjeldahl method [3]. Total available P (w/v) and K (w/v) were analyzed by extracting 5 g soil with 50 ml 2 M KCl, 100 ml 0.5 M NaHCO3, and 50 ml 1 M NH4OAc, respectively [35]. Microbial biomass C was determined by chloroform fumigation extraction method, using 0.5 M K2SO4 as extractant [22]. Soil was preconditioned by spreading between the two polythene sheets for an overnight period. It was then transferred to polythene bags and incubated for 7 days at 25°C in an air-tight container which contained two vials, one with 20 ml distilled water to maintain 100% relative humidity and other with soda lime to absorb CO2. The cover of the container was opened for a few minutes every day for the aeration. The soil was taken out after 1 week and mixed thoroughly for analysis of microbial biomass C by the fumigation extraction method as described above.
Cultivable Microbial Population
The microbiota in CCL, OCL, and FGL soils was determined by the culture enrichment technique [32]. The total microbial counts were determined using nutrient agar for bacteria, Kenknight and Munaier’s medium for Actinomycetes spp., and Rose Bengal Chloramphenicol agar (HI-MEDIA Laboratories Pvt., Ltd., Bombay, India) for selective isolation of fungi.
Microbial Metabolic Diversity Using Biolog
Biolog ECO GN2 and MT plates (Biolog, Inc., Hayward, CA, USA) were used to determine the community level functional profiling based on carbon source utilization pattern for CCL, FGL, and OCL soil samples. The Biolog MT plates were prepared using the manufacturer’s instructions (Biolog Inc., Hayward, CA 94545, USA). Individual 1-g soil samples were shaken in 9 ml of sterile saline MQW (0.85% NaCl) for 2 h and then made up to a final dilution of 10−2. After incubation, 150 μl of the sample was inoculated in each well of Biolog Eco and MT plates and incubated at 30°C. The rate of utilization is indicated by the reduction of tetrazolium, a redox indicator dye, which changes from colorless to purple. Data were recorded for day 1–14 at 590 nm. Microbial activity in each microplate expressed as average well color development (AWCD) was determined as described by Garland [12]. Diversity and evenness indexes and principal component analysis (PCA) was performed on the seventh day data divided by the AWCD [11]. Statistical analyses were performed using SPSS 16.0 and Statistica 7.0.
DNA Extraction and Pyrosequencing
Ultra Clean™ Soil DNA Isolation Kit (MoBio, Solana Beach, CA, USA) was used to isolate the DNA from CCL, FGL, and OCL soil samples according to the manufacturer’s instructions. Variable region V1-V2 for 16S rRNA gene was amplified by using 27 F and 338R composite primers [8, 48]. Unique 10 base identifiers (AGACTATACT for CCL, AGCGTCGTCT for FGL and AGTACGCTAT for OCL, respectively) were used to tag PCR product.
Replicate PCR reactions were performed for each sample; reaction mixture consists of 1 μl (10 pmol/μl) of each primer, 40 ng of DNA, 1 μl of dNTPs mix (10 mM stock), 1 unit of Phusion hot start II high-fidelity DNA polymerase, and 10 μl of 5× Phusion HF buffer. Final volume of reaction was adjusted to 50 μl by adding PCR-grade water. Amplification was performed using an initial denaturation of 3 min at 98°C followed by 30 cycles, denaturation at 98°C for 10 s, annealing at 55°C for 20 s, and elongation at 72°C for 30 s. Final extension was at 72°C for 10 min. Amplified products were run on agarose gel; specific bands were excised and amplicons were purified by Qiagen gel purification kit. Concentrations of eluted and purified amplicons were measured by Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Equal amount of PCR products were mixed in a single tube and sequenced using Roche 454 titanium chemistry.
Analysis of Sequences
After pyrosequencing, all sequence data were screened and filtered for quality and length using PERL script [13]. Sequences were trimmed and binned by samples using the unique 10 base identifiers as stated above. Sequences failing following quality criterion like not having perfect match with 10 base identifier sequences or specific primers were excluded for further analysis: less than 200-bases long reads, quality score less than 20, having any ambiguous base, and sequences with more than six homopolymers. Subsequently, chimeric sequences were removed by MOTHUR chimera slayer command [38]. Sequences were classified by RDP classifier tool [49] with 50% confidence threshold as recommended for short sequences. Sequences were grouped into species- (≥97% similarity), genera- (≥95% similarity), family- (≥90% similarity), and phylum (≥80%)-level phylotypes [39] by using MOTHUR [40]. Shannon and Simpson diversity indices and shared phylotypes were also calculated by MOTHUR [40] at every level (species to phylum) of phylotypes. Sequence libraries were compared pairwise by RDP Library compare tool [49]. Over all differences in bacterial composition in three libraries were tested by ∫-Libshuff implemented in MOTHUR [40]. The two libraries were considered to be significantly different from each other if the lower of the two P values generated by ∫-Libshuff was below or equal to the critical P value. The two libraries were considered to be significantly different from each other if the lower of the two P values generated by ∫-Libshuff was below or equal to the critical P value [39].
Results
Physicochemical and Microbial Analysis
In order to analyze the influence of fertilizers fortification on soil physicochemical properties, microbial population and enzymatic activities are summarized in Table 1. Soil samples from three plots were deep black granular sandy loam in CCL, dark brown granular sandy loam from FGL, and black granular sandy loam from OCL. Results of this study demonstrate that total organic C content in OCL soil was about 2.71 and 2.19 times higher compared to the CCL and FGL soils, respectively. Microbial population (heterogeneous bacteria and actinomycetes) was also significantly higher in OCL compared to CCL and FGL soil which was further confirmed by higher microbial biomass carbon content in OCL soil which was approx. 1.5 times higher compared to CCL and FGL soils (Table 1). High inputs of cow manure and crop residues in the OCL resulted in significantly enhanced total available nitrogen, phosphorus, and potassium compared with FGL and CCL soils (Table 1).
Community Level Physiological Profiling of CCL, FGL, and OCL Soil Samples
Functional diversity and community structure of CCL, FGL, and OCL soils were measured by Biolog ECO and GN2 plates. Based on carbon source utilization pattern, Shannon, McIntosh, and Simpson diversity indices and related evenness were calculated, and results demonstrate that the highest diversity and evenness were recorded in OCL soil samples followed by CCL and FGL (Table 2). Results from categorized carbon source utilization pattern clearly demonstrate that amino acids, carbohydrates, and polymers were maximum utilized by OCL, while carboxylic acids were efficiently utilized by CCL soil. Amines and amides were utilized equally by CCL and OCL, whereas FGL utilizes miscellaneous carbon substrates more efficiently among other categories (Fig. 1). Data from Biolog MT plates for specific carbon source utilization pattern demonstrated that pectin, chitin, cellulose, starch, and phytic acid were utilized significantly higher by microflora of FGL as compared to CCL and OCL soil samples, whereas urea, lignin, p-nitrophenol, and glutamine as carbon substrate were utilized significantly higher by OCL as compared to FGL and CCL soil samples (Fig. 2). Principal component analysis was performed on carbon source utilization pattern of CCL, FGL, and OCL soil samples to describe the characteristics of samples which will help to understand their distribution and clustering. In clustering among the three soil samples, they were distributed separately among each other at 59.89% and 21.76% on the PCA vectors 1 and 2 axes (Fig. 3). FGL and CCL soil are closer to each other on this axis, while OCL was very distinct, indicating marked differences in the bacterial community structure.

Categorized carbon substrate utilization pattern of soil samples from chemical cultivated (CCL), fallow grass land (FGL), and organically cultivated land (OCL) using Biolog Eco and GN2 plates after 7-day incubation at 28°C. Bars with the same letter are not significantly different (at P < 0.05) using Waller Duncan test

Additional 11 substrate utilization patterns of soil samples from chemical cultivated (CCL), fallow grass land (FGL), and organically cultivated land (OCL) using Biolog in MT plates after 7-day incubation at 28°C. Bars with the same letter are not significantly different (at P < 0.05) using Waller Duncan test

Principal component analysis based on carbon source utilization pattern by CCL, FGL, and OCL soil bacterial communities after 7-day incubation at 28°C. A total of 126 carbon substrate from Biolog Eco and GN2 (31 and 95 carbon substrate) were used in analysis
Phylogenetic Assignment and Bacterial Community Structure of CCL, FGL, and OCL Soil Samples
Pyrosequencing results of 16S rRNA gene sequencing were used to assign the phylogenetic position and community structure for CCL, FGL, and OCL soil samples. For our present study, we have used the V1-V2 region of 16S rRNA gene to evaluate the bacterial community structure by using high throughput pyrosequencing. A total of 20,132 sequences were obtained for three soil samples, after quality control of 16,608 sequences were recovered belongs to 6,379, 4,835, and 5,394 sequences for CCL, OCL, and FGL, respectively. Among the three samples, these sequences were classified in 15 known phylum and distributed as Proteobacteria (29.8%), Acidobacteria (22.7%), Actinobacteria (11.1%), Bacteroidetes (4.7%), Gemmatimonadetes (3.4%), Firmicutes (1.1%), Chloroflexi (0.8%), Cyanobacteria (0.6%), TM7 (0.4%), Nitrospira (0.4%), Verrucomicrobia (0.3%), WS3 (0.2%), OP10 (0.2%), Planctomycetes (0.1%), and OD1 (0.01%), while 24.3% sequences were unclassified using Ribosomal database (http://rdp.cme.msu.edu/classifier/classifier.jsp). Most abundant phylum Proteobacteria is further distributed in five subphylum Alphaproteobacteria (40.7%), Betaproteobacteria (23.0%), Deltaproteobacteria (11.7%), Gammaproteobacteria (11.2%), and unclassified Proteobacteria (13.3%).
To compare the taxonomic differences among the CCL, OCL, and FGL soil samples, 16S rRNA gene libraries were pairwise compared by RDP library compare tool. This tool combines a rapid taxonomic classifier (Naïve Bayesian classification) with a statistical test to flag taxa significantly different between two libraries. Gram-negative phyla Proteobacteria (30.1%, 35.1%, and 24.17% in CCL, OCL, and FGL, respectively), Bacteroidetes (4.7%, 6.1%, and 3.1% in CCL, OCL, and FGL, respectively), and Gemmatimonadetes (2.4%, 5.4%, and 2.4% in CCL, OCL, and FGL, respectively) were found to be significantly abundant (P < 0.01) in OCL compared to FGL and CCL soils. On the other hand, Actinobacteria were significantly abundant (P < 0.01) in CCL in comparison to FGL or OCL soil. About 18.3% Actinobacteria were observed in CCL soil, while in FGL and OCL, presence was only 12.0% and 2.9%, respectively (Fig. 4). Other main phyla Acidobacteria was found to be significantly abundant in FGL soil (30.0%) in comparison to CCL (17.8%) and OCL (20.5%). Significant differences in other minor phyla like TM7, Chloroflexi, Firmicutes, Nitrospira, Planctomycetes, Cyanobacteria, and OD1 were also observed.

Phylogenetic classification of relative abundance of dominant bacterial phyla in CCL, FGL, and OCL soils obtained from a ribosomal database project classifier analyses
Summary and Diversity Measures of CCL, FGL, and OCL Soil Samples
Rarefaction analysis of bacterial communities derived from CCL, FGL, and OCL soil samples are depicted at 3% (species), 5% (genera), 10% (family), and 20% (phylum) dissimilarity in Fig. 5. At 20% genetic distance, almost all curves showed saturation, indicating that the surveying effort covered almost the full extent of taxonomic diversity at phylum level while at family, genera, and species level we need more sequencing efforts. At species level, maximum number 2,389 operational taxonomic units (OTUs) were recorded from OCL soil samples followed by 2,243 and 2,233 for CCL and FGL samples, respectively. Rarefaction of the soil samples did not reach saturation level for species, genera, and family even after retrieval of more than 4,800 sequences. Good’s query coverage among these samples ranges 68.7–80.6% for species, 78.2–87.7% for genera, 92.3–96.2% for family, and 99.3–99.7% for phylum level. Variation in Good’s query coverage may be due to the number of sequences obtained from individual sample, and number of sequences at any given phylogenetic level is strongly affected by the number of sequences analyzed [39]. Simpson diversity index were recorded highest in OCL samples followed by CCL and FGL at all calculated phylogenetic levels. Shannon diversity index, species observed (S obs), non-parametric estimators of diversity (Chao1), and abundance (Ace) values have not shown similar pattern as obtained by number of OTUs and Simpson diversity index (Table 4).

Rarefaction analysis based on V1-V3 16S rRNA pyrosequencing data depicting at species (3%), genera (5%), family (10%), and phylum (20%) level for three soil samples from chemical cultivated (CCL), fallow grass land (FGL), and organically cultivated land (OCL)
Comparison of Bacterial Community Structure of CCL, FGL and OCL Soil Samples
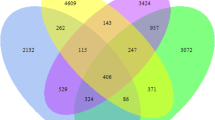
∫-Libshuff analysis was performed in order to estimate the differences or similarities among the community structure of CCL, FGL, and OCL soil samples. Pairwise comparison of these libraries was found to be significantly different (P < 0.0001) from each other in both the direction (X vs. Y and Y vs. X) and results are summarized in Table 3. Shared species among the CCL, FGL, and OCL soil samples were represented by Venn diagram, and results clearly demonstrated that 302 species were common for all three soil samples at 0.03 distances, while there were 361 species common for CCL and FGL, 159 species for FGL and OGL, and 332 for OCL and CCL (Fig. 6).

Venn diagram (at distance 0.03) showing shared and unique genera identified in 16S rRNA gene sequences recovered from bar-coded pyrosequencing reads of soil samples from chemical cultivated land (CCL), fallow grass land (FGL), and organically cultivated land (OCL)
Discussion
Community-level physiological profiling and 16S rRNA gene-based approaches have been used extensively to study the microbial communities in various habitats [21, 30, 32, 37, 42, 46]. In present study, we have showed that compared to OCL or FGL soil samples, microbial activities and diversity are significantly reduced in chemical fertilizer-managed soil (CCL). Organic manure has significant impact on the biomass C and the activity, compared with mineral fertilizers [4]. Application of organic amendments to soil not only increases the total organic carbon content and its different fractions but also has a series of effects on microbial proliferation and activity [2]. Several studies have reported that enzyme activities in the soil are related to the organic matter and microbial biomass [25, 31, 44].
The application of cow dung to improve the crop yield has been indicated since the time of Kautilya (c. 300 BC), which was later continuously followed by Indian farmers in various ways [34]. Swain and Ray [47] has reported some plant growth promoting rhizobacteria isolated from cow dung; our result supported the view that cow dung compost amendment (OCL) activates comparatively more microorganisms than conventionally used chemical fertilizers. This higher activity of microbes possibly leads to the enhanced soil organic carbon, soil organic nitrogen, and microbial biomass in OCL and FGL soil compared to CCL as shown in Table 1.
We observed a collapse of microbial functional diversity in CCL soil compared to OCL, which is in accordance with earlier findings, showing higher biodiversity and fertility in organic plots [28]. In recent study, our group have also shown shift in functional microbial diversity, during the conversion of FGL to OCL, and the improvement of soil health due the enhancement of microbial functional diversity by using CLPP approach [32]. Biolog-based functional diversity and evenness indices were reported higher in OCL as compared to FGL and CCL soil samples. Principal component analysis based on carbon source utilization pattern of CCL, FGL, and OCL soil samples clearly reflect that functional diversity in CCL and FGL was more similar as compared to OCL. Earlier also, we have reported that organically cultivated land compared with fallow grass land soil has significantly higher diversity indices and evenness [32].
We compared 16S rRNA gene sequences generated by high throughput pyrosequencing; this technique provides a deeper insight of microbial community description with a high level of accuracy (Table 4) [9, 18]. Soil bacterial diversity is enormously high, and most of the bacterial taxa are rare and may represents up to 37% of total bacterial load [6]. As expected rarefaction curves did not reached plateau at any level of phylotypes, although analyzed datasets consist of minimum of 4,800 sequences per samples in this study. Lauber et al. [26] also observed the similar pattern with about 1,000–2,000 sequences per soil samples.
In our study, we have observed diminished microbial diversity in OCL soil in 16S rRNA gene-based analysis. Earlier pyrosequencing technique has revealed that microbial diversity is under the influence of the nature of ecosystem; soil from agricultural land and forest has different bacterial diversity whereas soil contaminated with heavy metal has reduced bacterial diversity [10]. Recently, Wu et al. [50] demonstrated that abundance and composition of bacterial communities were not sensitive to inorganic fertilization while significantly affected by applying inorganic fertilizers with rice straw by using T-RFLP and 16S rRNA clone library. Similar to these results in the present study, direct negative effect of chemical fertilizer on bacterial diversity was observed and differences in observations are may be likely due to the different methodological approach and relatively deep mining of bacterial communities using 454 pyrosequencing.
In addition to reduced bacterial functional and phylogenetic diversity, a distinct shift of Gram-negative and Gram-positive bacterial phyla was observed in CCL, FGL, and OCL soil samples. The higher abundance of Gram-negative phyla Proteobacteria, Bacteroidetes, and Gemmatimonadetes was recorded in OCL soil samples as compared to FGL and CCL; similar to these results, abundance of Gram-negative bacterial phyla were reported higher in control soil (where no chemical fertilizer was amended) and compost-amended soil in comparison to chemical fertilizer-treated soil has been shown by fatty acid methyl ester and phospholipid fatty acid analysis [19, 36]. Observed higher abundance of Proteobacteria, Bacteroidetes, and Gemmatimonadetes is expected to result in enhanced cycling of essential micro or macro nutrients, which may partially improve soil fertility and plant growth efficiency [27]. The higher abundance of Acidobacteria could be explained by the recent results of Jones et al. [23] which clearly demonstrated that pH is strongly correlated with the abundance of Acidobacteria. Similar to this study, in our results, also FGL soil comprises higher abundance of Acidobacteria followed by OCL and CCL which are correlated well with lower soil pH compared with other treatments. We have also observed higher level of Actinobacteria in CCL soil as compared to other two treatments in our investigation, supporting the findings of Aira et al. [1] showing lower Gram-positive bacterial biomass in rhizosphere of maize plants in the plots treated with inorganic fertilizer than in those treated with organic fertilizers.
The presented study demonstrated that the microbial community structure, diversity, and bacterial phylogeny in CCL, OCL, and FGL soil samples have been practiced in the last 16 years. Pyrosequencing revealed significant changes in bacterial phyla distribution among the mentioned soil types. Predominant bacterial phylum were identified under the organically cultivated soil (i.e., Proteobacteria, Bacteroidetes, and Gemmatimonadetes), fallow grassland (i.e., Acidobacteria), and in chemically treated soil (i.e., Actinobacteria) which may imply that these bacteria play major roles in soil processes under these different management systems. This study also demonstrated that organic amendment can enhance the population of certain bacterial phyla which can be correlated with other soil properties such as pH, total organic C, total organic N, and microbial biomass C. Uncovering of previously unknown bacterial populations was able to present a comprehensive view of the shifts in the bacterial community structure of different land management practices for long term.
References
Aira M, Brandón MG, Lazcano C, Bååth E, Domínguez J (2010) Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol Biochem 42:2276
Bastida F, Kandeler E, Moreno JL, Ros M, García C, Hernández T (2008) Application of fresh and composted organic wastes modifies structure, size and activity of soil microbial community under semiarid climate. Appl Soil Ecol 40:318
Bremner JM, Mulvaney CS (1982) Nitrogen-total. In: Page AL (ed) Methods of soil analysis. Part 2. Chemical and microbiological properties. Book series no. 9 SSSA, Madison, p 595
Chu H, Fujii T, Morimoto S, Lin X, Yagi K, Hu J, Zhang J (2007) Soil microbial biomass, dehydrogenase activity, bacterial community structure in response to long-term fertilizer management. Soil Biol Biochem 39:2971
Dunfield KE, Germida JJ (2004) Impact of genetically modified crops on soil and plant-associated microbial communities. J Environ Qual 33:806
Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, Sukharnikov LO, Roe BA, Davis JP, Schloss PD, Bailey VL, Krumholz LR (2008) Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol 74:5422
Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626
Fierer N, Hamadyc M, Lauberb CL, Knightd R (2008) The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 18:17994
Fulthorpe RR, Roesch LFW, Riva A, Triplett EW (2008) Distantly sampled soils carry few species in common. ISME J 2:901
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveals great bacterial diversity and high metal toxicity in soil. Science 309:1387
Garland JL, Mills AL (1991) Classification and characterization of heterotrophic microbial communities on the basis of patterns of community level sole carbon source utilization. Appl Environ Microbiol 57:2351
Garland JL (1996) Analytical approaches to the characterization of samples of microbial communities using patterns of potential C source utilization. Soil Biol Biochem 28:213
Giongo A, Crabb DB, Davis-Richardson AG, Chauliac D, Mobberley JM, Gano KA, Mukherjee N, Casella G, Roesch LF, Walts B, Riva A, King G, Triplett EW (2010) PANGEA: pipeline for analysis of next generation amplicons. ISME J 4:852
Gomez E, Ferreras L, Toresani S (2006) Soil bacterial functional diversity as influenced by organic amendment application. Biores Technol 97:1484
Goyal S, Mishra MM, Hooda IS, Singh R (1992) Organic matter–microbial biomass relationships in field experiments under tropical conditions: effects of inorganic fertilization and organic amendments. Soil Biol Biochem 24:1081
Hallmann J, Rodríguez-Kábana R, Kloepper JW (1999) Chitin-mediated changes in bacterial communities of the soil, rhizosphere and within roots of cotton in relation to nematode control. Soil Biol Biochem 31:551
Heilmann B, Lebuhn M, Beese F (1995) Methods for the investigation of metabolic activities and shifts in the microbial community in a soil treated with a fungicide. Biol Fertil Soils 19:186
Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Welch DM, Relman DA, Sogin ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4:e1000255
Islam MR, Trivedi P, Palaniappan P, Reddy MS, Sa T (2009) Evaluating the effect of fertilizer application on soil microbial community structure in rice based cropping system using fatty acid methyl esters (FAME) analysis. World J Microbiol Biotechnol 25:1115
Islam MR, Trivedi P, Madhaiyan M, Seshadri S, Lee G, Yang J, Kim Y, Kim M, Han GH, Chauhan PS, Sa T (2010) Isolation, enumeration, and characterization of diazotrophic bacteria from paddy soil sample under long-term fertilizer management experiment. Biol Fertil Soils 46:261
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719
Jenkinson DS, Powlson DS (1976) The effects of biocidal treatment on metabolism in soil. I. Fumigation with chloroform. Soil Biol Biochem 8:167
Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N (2009) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3:442e453
Kinyua D, McGeoch LE, Georgiadis N, Young TP (2010) Short-term and long-term effects of soil ripping, seeding, and fertilization on the restoration of a tropical Rangeland. Rest Ecol 18:226–233
Lagomarsino A, Moscatelli MC, Di Tizio A, Mancinelli R, Grego S, Marinari S (2009) Soil biochemical indicators as a tool to assess the short-term impact of agricultural management on changes in organic C in a Mediterranean environment. Ecol Indic 9:518
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil ph as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111
Lesaulnier C, Papamichail D, McCorkle S, Ollivier B, Skiena S, Taghavi S, Zak D, van der Lelie D (2008) Elevated atmospheric CO2 affects soil microbial diversity associated with trembling aspen. Environ Microbiol 10:926
Maeder P, Fliessbach A, Dubois D, Gunst L, Fried P, Niggli U (2002) Soil fertility and biodiversity in organic farming. Science 296:1694
Marinari S, Mancinelli R, Campiglia E, Grego S (2006) Chemical and biological indicators of soil quality in organic and conventional farming systems in Central Italy. Ecol Indic 6:701
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 6:e17000
Nannipieri P, Ascher J, Ceccherini MT, Landi L, Pietramellara G, Renella G, Valori F (2008) Effects of root exudates in microbial diversity and activity in rhizosphere soils. In: Nautiyal CS, Dion P (eds) Molecular mechanisms of plant and microbe coexistence. Soil Biology Series, vol 15. Springer, Berlin, p 339
Nautiyal CS, Chauhan PS, Bhatia CR (2010) Changes in soil physico-chemical properties and microbial functional diversity due to 14 years of conversion of grassland to organic agriculture in semi-arid agroecosystem. Soil Till Res 109:55–60
Nelson DW, Sommers LE (1982) Total carbon, organic carbon, and organic matter. In: PageMiller AL, Keeney RH (eds) Methods of soil analysis. Part 2, 2nd edn. American Society of Agronomy–Soil Science Society of America, Madison, p 539
Nene YL (2006) Indian pulses through the millennia. Asian Agri Hist 10:179–202
Page AL, Miller RH, Keeney DR (1982) Methods of soil analysis. Part 2, Chemical and microbiological properties, 2nd ed., Agronomy vol. 9. ASA, SSSA, Madison, p 1159
Peacock A, Mullen M, Ringelberg D, Tyler D, Hedrick D, Gale P, White DC (2001) Soil microbial community responses to dairy manure or ammonium nitrate applications. Soil Biol Biochem 33:1011
Sala MM, Terrado R, Lovejoy C, Unrein F, Pedrós-Alió C (2008) Metabolic diversity of heterotrophic bacterioplankton over winter and spring in the coastal Arctic Ocean. Environ Microbiol 10:942
Schloss PD, Larget BR, Handelsman J (2004) Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 70:5485–5492
Schloss PD, Handelsman J (2004) Status of the microbial census. Microbiol Mol Biol Rev 68:686
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537
Schönfeld J, Gelsomino A, Overbeek LS, Gorissen A, Smalla K, Elsas JD (2003) Effects of compost addition and simulated solarisation on the fate of Ralstonia solanacearum biovar 2 and indigenous bacteria in soil. FEMS Microbiol Ecol 43:63
Shannon D, Sen AM, Johnson DB (2002) A comparative study of the microbiology of soils managed under organic and conventional regimes. Soil Use Manag 18:274
Sheik CS, Beasley WH, Elshahed MS, Zhou X, Luo Y, Krumholz LR (2011) Effect of warming and drought on grassland microbial communities. ISME J 5:1692
Shi W, Dell E, Bowman D, Iyyemperumal K (2006) Soil enzyme activities and organic matter composition in a turfgrass chronosequence. Plant Soil 288:285
Shinjiro K, Susumu A, Yasuo T (1988) Effect of fertilizer and manure application on microbial numbers, biomass, and enzyme activities in volcanic ash soils: I. Microbial numbers and biomass carbon. Soil Sci Plant Nutr 34:429
Staddon WJ, Duchesne LC, Trevors JT (1997) Microbial diversity and community structure of post-disturbance forest soils as determined by sole-carbon-source utilization patterns. Microb Ecol 34:125
Swain MR, Ray RC (2009) Biocontrol and other beneficial activities of Bacillus subtilis isolated from cow dung microflora. Microbiol Res 164:12
Torsvik V, Øvreås L (2002) Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol 5:240
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261
Wu M, Qin H, Chen Z, Wu J, Wei W (2011) Effect of long-term fertilization on bacterial composition in rice paddy soil. Biol Fertil Soils 47:397
Wu T, Chellemi DO, Graham JH, Martin KJ, Rosskopf EN (2008) Comparison of soil bacterial communities under diverse agricultural land management and crop production practices. Microb Ecol 55:293
Zak DR, Holmes WE, White DC, Peacock AD, Tilman D (2003) Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84:2042
Zhong WH, Cai ZC (2007) Long-term effects of inorganic fertilizers on microbial biomass and community functional diversity in a paddy soil derived from quaternary red clay. Appl Soil Ecol 36:84
Acknowledgments
We are indebted to Dr. D.V. Amla, Chief Scientist, CSIR-National Botanical Research Institute, Lucknow, India for his valuable suggestions and critical editing of the manuscript. The study was partially supported by TATA Innovation Fellowship, Department of Biotechnology, Government of India, New Delhi and Task Force grant NWP-006 from Council of Scientific and Industrial Research (CSIR), New Delhi and awarded to CSN. VC would like to thank CSIR for providing her Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chaudhry, V., Rehman, A., Mishra, A. et al. Changes in Bacterial Community Structure of Agricultural Land Due to Long-Term Organic and Chemical Amendments. Microb Ecol 64, 450–460 (2012). https://doi.org/10.1007/s00248-012-0025-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-012-0025-y