
Abstract
In this study, we employed pyrosequencing-based analysis of the V4-V6 16S rRNA gene region and community level physiological profiling (CLPP) to characterize and compare bacterial community composition and metabolic activities in order to determine if microbial composition and activity were able to recover in land transitioning from agriculture to natural wetland. The sample sites included natural wetland (with native vegetation), reclaimed for tillage (agriculture), and abandoned tilled (wetland restoration) soils. The entire data set comprised 78,790 good-quality sequences, 62,364 of which were classified in 23 known phyla. The dominant taxonomic groups presented in all samples were Proteobacteria (34.3%), Acidobacteria (15.5%), Verrucomicrobia (6.9%), Chloroflexi (6.3%), Nitrospira (2.6%) and Actinobacteria (2.4%), while 27.1% of the sequences could not be classified to any known phylum. Microbial community composition and activity were found to cluster into two distinct groupings: one associated with agriculture and the other with restored and natural wetlands. The average well colour development (AWCD) based on carbon source utilization pattern was significantly higher in natural and restored wetland soils than in reclaimed soils, suggesting an improvement of soil health. In conclusion, these findings indicated that microbial community composition and activity were affected by land use and may be used to assess the extent of recovery of natural microbial-driven processes during wetland restoration. Furthermore, restored soil microbial communities in this study appear to be tended to a gradual return to those of natural wetland soils with time.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Microorganisms are one of the primary active components of soil. Their physiological activity performs vital ecosystem functions that include element cycling, organic matter decomposition, and soil structure formation (Swift et al. 1979; Dominati et al. 2010; Zhao et al. 2013). Microbial community composition and activity are greatly influenced by an assortment of biotic and abiotic factors including plant species richness, pH, nutrient availability, and soil organic carbon (Zak et al. 2003; Fierer and Jackson 2006; Nautiyal et al. 2010). Furthermore, differences in rates and diversity of microbial activity have also been linked to changes in community composition (Castro et al. 2010; Fierer et al. 2012). Land use changes often result in modification of vegetation cover and soil physicochemical properties, which, in turn, influence microbial-driven ecological processes (Bending et al. 2002; Bisset et al. 2011). Hence, understanding the links among land use, microbial community composition and activity is an active and vital topic of contemporary microbial ecology research (Fierer et al. 2013; Carbonetto et al. 2014).
Microbial community composition (van Dijk et al. 2009; Kubler et al. 2014) or activity (Bending et al. 2002; Lewis et al. 2010) has been documented as an indicator of ecosystem responses to anthropogenic alteration and ecological recovery (Harris 2003). However, previous studies have shown that monitoring both microbial composition and activity in conjunction with different types of land use mainly associated with various agricultural practices (Chaudhry et al. 2012; Yu et al. 2013). We extended the scope of work that has been done in previous studies by comparing microbial community composition and activity in undisturbed wetland, agricultural land converted from natural wetland, and restored wetland habitat.
Freshwater wetlands provide habitat and food for waterfowl and other wildlife, protect shorelines, and improve water quality of rivers and lakes, etc. (Richardson and Marshall 1986; Roulet 2000; Alewell et al. 2008). In addition, wetlands play a crucial role in sequestering carbon as they store 25–30% of the world’s carbon pool despite only covering 5–8% of the land surface (Mitsch and Gosselink 2007). Despite the critical ecological function that wetlands provide, 50% of the world wetlands have been destroyed in the last century (World Conservation Union 1996). This is mainly attributed to anthropogenic activities, such as reclamation for agricultural use, wetland drainage and in-filling, and deforestation.
Poyang Lake is the largest freshwater lake in China. In this basin, wetland degradation for agricultural purposes is significant. Lake capacity, primarily its wetland areas, decreased by 22% from 37 to 28.9 billion m3 during a 44-year period from 1954 to 1998 (Min 1999). Fortunately, overuse and reclamation of the Poyang Lake wetland has decreased since the 1980s as the government realized the serious consequences of destroyed wetlands and implemented a policy of restoring wetland habitat by abandoning agricultural land.
In this study, our aims were to (i) determine if microbial composition and activity would vary in natural, restored, or reclaimed wetlands; (ii) assess the extent of recovery for microbial composition and activity associated with wetland restoration. To achieve these objectives, the structure of microbial community was analyzed by 454 pyrosequencing of the 16S rRNA gene, and microbial activity was determined using community level physiological profiling (CLPP). The use of 454 pyrosequencing was selected due to its well-known capacity to accurately elucidate the characters of microbial communities (Quince et al. 2009; Zhang et al. 2012; Lu et al. 2015).
Material and Methods
Site Description and Experimental Design
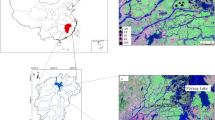
The studied sites were located on JiangXiang island in the southwest corner of Poyang Lake (Jiangxi Province, China, 28°45′N,116°01′E) (Fig. 1). This area belongs to a subtropical monsoon climate zone with significant variations in both precipitation and temperature. It has an annual average temperature of 17.5 °C.The average annual precipitation is 1470 mm, and 69.4% of the annual precipitation occurs during April–September. The soil type of the experimental field is classified as semi-hydromorphic soils according to the Chinese Soil Taxonomy.

Poyang Lake basin and distribution of samples locations
In our study, there were four different land use types located in the adjacent areas: natural wetland (NW), 38-year long reclaimed field (RC38), 48-year long reclaimed field (RC48) and 25-year long restored wetland (RS25). The NW and RS25 have loamy clay texture, while RC38 and RC48 have clay loam texture (Table S1). The NW is now mainly covered with Carex cinerascens and Phalaris arundinacea without artificial control or management. Two reclaimed fields, RC38 and RC48, have been converted from natural wetland for rice (Oryza sativa) cultivation since 38 years and 48 years ago, respectively. On the natural wetland, embankments built along the lake hold back the water and the fields were plowed for rice cultivation. Along with irrigation and fertilization, intensive cultivation of the land by double rice cropping a year are undergoing for both two reclaimed fields. In each cropping season, compound fertilizer (N, K, P, Ca) were applied before planting, and urea was spread during the period of last phase of flowering and early filling of rice. The RS25, a previously reclaimed filed for rice cultivation, where embankments were destroyed and has been left unplanted for 25 years. Now it has been undergoing natural restoration and mainly covered by C. cinerascen. For both natural and restored wetlands, there is a dynamic wetland system where water level is generally rising and vegetation is completely inundated during the summer rainy season, while water level is lowered below soil surface layer during winter dry season.
Soil Sampling and Chemical Analysis
Soil samples were collected in May 2011 when both rice in reclaimed fields and vegetation in restored and natural wetlands were actively growing. During the soil sample collection, all sites under different land use types have a generally identical water level. The purpose of collecting samples in this season was to ensure that comparisons made between land use types were not biased by hydrology. The depth of the topsoil horizons at four studied sites is 20 cm. Four similar 20 m × 20 m plots with 100 m apart from each other were selected for each land use type and in each plot, topsoil samples were collected from five randomly selected soil cores (2.5 cm in diameter; 20 cm in depth) and mixed to give one composite sample (US Environmental Protection Agency 2014). The plots for each land use type were separated by the rice field ridges or walkways. Therefore, there were a total of 16 soil samples in this study. After roots and organic debris in the soil samples were removed by hand, each soil sample was passed through a 2 mm sieve, homogenized, and then divided into two subsamples. One subsample was stored at −80 °C for the determination of microbial community composition and activity, and the other subsample was air-dried for analyses of soil physicochemical properties.
Soil total organic C (TOC), total N (TN) and total P (TP) were evaluated by the K2CrO7/H2SO4 oxidation procedure, the microkjeldahl method and the phosphomolybdic acid blue color method, respectively (Lu 2000). Soil pH was measured by a pH Meter (FE20K, Mettler) at a soil: water ratio of 1:2.5 (w/v). Soil moisture content was measured by weighing after oven-drying at 75 °C for 24 h. Soil bulk density was determined by the clod method (Blake 1965). The bulk density of soil clods was calculated from their mass and volume. The volume was determined by coating the clod with melted paraffin and by weighing it first in air, then again while immersed in water. Soil texture was determined by the Bouyoucos hydrometer method (Bouyoucos 1962).
Community-Level Physiological Profiles
Community–level physiological profile patterns (CLPP) of soil bacterial communities were assessed by the BIOLOG procedure, using BIOLOG 96-well Eco-Microplates (Biolog, Inc., Hayward, CA) with 31 different carbon sources (Garland and Mills 1991; Sharma et al. 1998; Girvan et al. 2003; Bisset et al. 2011). Briefly, moist soil samples with a unified dry weight (10 g) was extracted with 100 ml sterile saline solution (0.85% NaCl) by shaking for 10 min then the suspension was diluted 100-fold. The unified dry weight (10 g) of soil samples was calculated from fresh weight and moisture content. Aliquots (150 μL) of the 1:1000 (w/v) soil suspensions were directly added to each of the 96 wells, and then the Microplates were incubated at 25 °C in the dark for 10 days. Colour development in the wells was read at 590 nm every 12 h using the MicroStation™ System (BIOLOG, USA). The average well colour development (AWCD) was used to express general microbial activity (Garland and Mills 1991).
DNA Extraction and Pyrosequencing
DNA of homogenized samples (0.5 g) was extracted using the PowerSoil DNA Isolation kit (MoBio, USA) according to manufacturer instructions. The V4-V6 region of the bacterial 16S rDNA was PCR amplified with 10-bp Bar-coded primers 530F (5′-GTGCCAGCMGCNGCGG-3′) and 1100R (5′-GGGTTNCGNTCGTTG-3′) and containing the A and B 454 FLX adapters. These primers were selected based on recent results indicating that this region is the best performing for profiling and identification of diverse members of bacterial communities (Turner et al. 1999; Acosta-Martínez et al. 2008; Lu et al. 2015). The PCR reactions were as previously described (Acosta-Martínez et al. 2008). Replicate PCR reactions were performed for each sample then pooled to minimize PCR bias, and finally established a single library. Following PCR, DNA amplicons were purified with ultraclean Gelspin DNA purification kit (Mobio) and quantified using Quant-iT Picrogreen dsDNA assay kit (Invitrogen). Amplicon pyrosequencing was performed using GS Junior Titanium chemistry at the Kent State University Biology Department. The pyrosequencing-derived 16S rDNA sequence datasets were submitted to the NCBI Sequence Read Achieve (SRA) under accession number SRP035329.
Organization of Pyrosequencing Data
Barcodes of the reads were scanned and trimmed using the Pipeline Initial Process on Ribosomal Database Project (RDP) (https://pyro.cme.msu.edu/init/form.spr). Then reads were filtered using Mothur software package. Poor quality reads, sequences shorter than 200 bp, and sequences identified as chimeras were removed (Schloss et al. 2009). The qualified sequences were clustered into operational taxonomic units (OTUs), which were defined by 97% sequence identity, using the RDP pyrosequencing pipeline (http://wildpigeon.cme.msu.edu/pyro/index.jsp) (Cole et al. 2004). Taxonomy was subsequently assigned to representative OTUs using the Greengenes database classifier at a confidence threshold of 80% (Wang et al. 2007). Each sample was randomly re-sampled and normalized, based on the sample with the least number of reads.
Statistical Analysis
In order to quantify and compare the distribution of community pyrosequencing data, several indices were calculated. Community richness was estimated with Chao1 (Gotelli and Colwell 2001), while Shannon and Simpson indices were used to compare community diversity. Differences in bacterial communities from four fields were tested by ∫-Libshuff implemented in Mothur (Schloss et al. 2009). The two libraries were considered to be significantly different from each other if the lower of the two P values generated by ∫-Libshuff was below or equal to the critical P value (Schloss and Handelsman 2004). A heat map comparing the relative abundance of phyla in each community was generated using the NCSS 2007 software.
Redundancy analysis (RDA; LeGendre and Legendre 1998) was conducted to determine if microbial activity (categorized carbon source utilization) was significantly correlated with microbial community composition. If any categorized carbon sources were significantly correlated with community composition, we wanted to determine if there were taxa that may be driving this pattern. In order to test that question, we used linear regression and Pearson correlation to determine if there were significant relationships between carbon source utilization and dominant genera (relative abundance > 1%) within communities. Likewise, potential correlations between the relative abundance of individual phyla and soil properties were examined by Person correlation coefficients.
We used one-way ANOVA to determine if there were significant differences in soil properties, microbial activities and community diversity (Shannon and Simpson indices) and richness (Chao1) associated with different land use types. Where the F values were significant, post hoc comparisons were made using the least significant difference test at the 0.05 probability level. Statistical analysis was performed using SPSS 16.0 and Statistica 7.0.
Results
Soil Chemical Properties
There were significant differences in soil chemical characteristics between reclaimed and natural wetland soils. The levels of TOC, TN and C/N ratio decreased in the following order: reclaimed > restored > natural wetland. Reclaimed for agriculture soils had significantly higher soil moisture, TOC, and TN than either the restored or natural wetland sites (P < 0.05, Table 1). TP contents were significantly higher in the RC38 soil than any other soil types (P < 0.05).
Microbial Activity
AWCD increased with the incubation time during the 10-day period (Fig. 2). Significant differences (P = 0.001) were found in the AWCD among reclaimed, restored, and natural wetland soils. The highest AWCD was in restored wetland soil, followed by natural wetland and reclaimed soil.

AWCD of the BIOLOG Eco plates for microbial samples from reclaimed (RC48 and RC38), restored (RS25) and natural wetland (NW) soils. RC48: 48-yr-old reclaimed soils; RC38: 38-yr-old reclaimed soils; RS25: 25-yr-old restored wetland soils; NW: natural wetland soils
Activity differences among the four communities were analyzed in detail with categorized carbon source utilization. Polymers (P = 0.001), carbohydrates (P = 0.011), and carboxylic acids (P = 0.020) were utilized most efficiently by microbes living in restored wetland soil (RS25), while amines, amino acids, and phenolic acids were utilized equally by microbes (P > 0.05) in all sites (Fig. 3).

Carbon source utilization patterns of soil samples from reclaimed (RC48 and RC38), restored (RS25) and natural wetland (NW) at 72 h absorbance. Bars with the same letter are not significantly different (P < 0.05)
Pyrosequencing, Richness and Diversity
A total of 62,364 reads were obtained from pyrosequencing after removal of low-quality and chimeric sequences (78,790 sequences before trimming) (Table 2). With a phylotype defined as those sequences sharing ≥ 97% identity, 1922 and 2838 OTUs were obtained from RC48 and RC38 samples, while 1814 and 1985 OTUs were recorded from RS25 and NW samples, respectively. Almost all rarefaction curves (Supplemental Fig. S1) reached saturation levels, indicating adequate sampling of actual species richness. The coverage among these samples is ranged from 88.7% to 96.3%. Richness and diversity were similar at all sites except RS25 which had the lowest Chao1 and Shannon indices (P < 0.0001) (Table 2).
Community Composition
After comparing the high quality sequences to the SILVA database, we classified the soil microbial community into known (23 Phyla, 201 genera) and unclassified phylotypes (Fig. 4). Proteobacteria (32.36% to 36.27%) was the most abundant phylum in every sampling site, followed by Acidobacteria, Verrucomicrobia, Chloroflexi, Nitrospira and Actinobacteria. These six phyla accounted for more than 68% of the total bacterial community in each sample, while 24.40% – 30.02% sequences were unclassified. It showed that the proportions of these known phyla varied among the four different soil libraries. For instance, Delta-proteobacteria, Nitrospira, and WS3 were more abundant in RS25 and NW soils compared to reclaimed soils. On the other hand, Verrucomicrobia and Gamma-proteobacteria were more abundant in reclaimed than restored or natural wetland soils. In addition, Gemmatimonadetes was more abundant in the NW soil than other soils, while Chloroflexi was less abundant in NW soil than other soils (P < 0.05).

Relative abundance of the major bacterial phyla present in reclaimed (RC48 and RC38), restored (RS25) and natural wetland (NW) soils
To further compare community taxonomic differences, 16S rRNA gene libraries were pairwise compared by the RDP library compare tool. Libshuff analysis indicated that every pairwise comparison of site community composition was found to be significantly different (P < 0.001) from each other (Table 3). The specific taxa driving differences in community composition among sites were shown in Fig. 5. Despite individual site differences, the top of Fig. 5 indicated hierarchical clustering among bacterial composition of RC48 and RC38 sites, and a separate clustering of RS25 and NW sites. Heat map analysis also implied that the similarity of bacterial community composition between RS38 and RS48 was higher than RS25 and NW.

Heat map analysis of soil bacterial community composition from reclaimed (RC48 and RC38), restored (RS25) and natural wetland (NW) samples at the phylum level
At genus level, 61.26% of the sequences were unclassified. Among classifiable genera, 118 and 147 genera were found in RC48 and RC38 soils, respectively. 88 genera were detected in restored soil (RS25), and 108 genera were obtained from the NW soil. The predominant genera in all samples mainly consisted of Gp1 (16.10%), Subdivision3_genera_incertae_sedis (15.97%), Geobacter (9.07%), Gp2 (5.68%), Anaeromyxobacter (5.41%), Gp7 (4.72%), Thermodesulfovibrio (4.32%), Gp3 (2.71%), Gp6 (2.62%), and Ktedonobacter (2.07%) (Table S2).
Relationships between Soil Characteristic, Microbial Composition and Activity
Pearson correlation coefficients revealed significantly positive correlations of TOC with Gamma-proteobacteria (P = 0.018) and Alpha-proteobacteria (P = 0.043), while negative correlations of TOC with Delta-proteobacteria (P = 0.009), Firmicutes (P = 0.022) and Nitrospira (P < 0.001) were found. Chloroflexi was positively correlated with TN (P < 0.001).
Redundancy analysis indicated that microbial activity involving carbohydrate and carboxylic acid utilization was significantly correlated with community composition (P = 0.046) (Supplemental Fig. S2). Furthermore, we tested carbohydrate and carboxylic acid utilization against the relative abundance of the dominant genera at each site. We found significant correlations between carbohydrate utilization and Spartobacteria (P = 0.019), carboxylic acid and Rhizomicrobium (P = 0.044), as well as carboxylic acid and Spartobacteria (P = 0.006). However, linear regressions were generated and the correlations were shown to be negative.
Discussion
Our study showed that differences in microbial community composition (reclaimed sites in one cluster, restored and natural wetland sites in the other cluster, Fig. 5) were associated with land use types. The influence of land use on community composition varied in other studies. Some studies showed a strong land use effect on bacterial communities (Chaudhry et al. 2012; Guan et al. 2013), while another study indicated that the bacterial composition of freshwater wetland samples from managed agriculture areas were similar to the bacterial communities from undisturbed wetland soils (Drenovsky et al. 2010). In our study, bacterial diversity of restored and natural wetland soils were lower than reclaimed soil, this finding was consistent with the results in other wetland ecosystems, where reference soils have the least diverse bacterial community (Hartman et al. 2008).
The utility of Biolog plates to assess differences in wetland microbial activities has been previously demonstrated in forested, floodplain wetlands (Alewell et al. 2008), and peatland restoration (Andersen et al. 2010). Our results indicated significantly higher microbial activity in the natural and restored wetland sites than the reclaimed sites (Fig. 2). As higher Biolog activity implies the improvement of soil health (Rampelotto et al. 2013), our findings suggested that the natural and restored soils have a higher degree of soil health than the reclaimed soils.
Our study demonstrated that natural and restored wetland sites have lower diversity within their community composition but higher microbial activity than reclaimed sites. In particular, the levels of microbial carbon utilization activity associated with polymers, carbohydrates, and carboxylic acids in restoration site were higher than those found at any other sites (Fig. 3). The relatively high functional activity may be related to the environmental factors in this site. The soil physicochemical total C and N were highest in reclaimed soils, followed by restored wetland soils and natural wetland. Reduced nutrient availability in the wetland sites may drive the soil microbial communities to develop an increased capability to acquire diverse carbon sources, which may be reflected in higher Biolog activity. The opposite dynamic may occur in reclaimed soil; increased nutrient levels may support copiotrophic bacteria to out-compete oligotrophic bacteria and decrease overall richness in these sites. Our results provided some evidence in supporting the work which showed that the increased microbial richness and diversity were associated with reduced microbial activity (Jousset et al. 2011; Becker et al. 2012). While it is not possible to determine if this is absolutely occurring with this data set, these results point toward an avenue of possible further investigation that may be useful in increasing our understanding of microbial composition and activity associated with ecological restoration.
Soil moisture followed a pattern that was similar to the soil physiochemical total C, N and C/N ratio. This is due to artificial irrigation in reclaimed soil. The soil total C, N, C/N ratio and moisture gradients suggested that reclamation leads to the increased nutrient levels and soil moisture, while wetland restoration generally represents a return to less fertile and less saturated soil conditions. Moreover, soil texture fractions of restored wetland appear to be similar to those of natural wetland. These findings provided evidence that during the process of ecological restoration, soil microbial community activity can shift in a similar trend as natural conditions. This dynamic has been demonstrated using Biolog plates during wetland (Alewell et al. 2008) as well as forest (Chaudhry et al. 2012; Doi and Ranamukhaarachchi 2013) and grassland (Li et al. 2012) restoration.
Despite significant differences in microbial composition and activity associated with land use types, none of the individual soil physiochemical properties significantly affected activity or community composition in this work. Most likely there are complex interactions among land use, nutrient levels, soil properties, and dominant plant communities that drive the significant differences in microbial activity and community composition. For example, although significant difference in overall microbial activity between wetland and reclaimed soil was found in this study, the utilization patterns for individual carbon sources were not distinct. To illustrate this point, the level of N-acquiring utilization activity (amino acid, and amine) in natural and restored wetland soil was even higher than that found in reclaimed soil. These sites had the lowest levels of N-associated physiochemical variable (TN), supporting the fact that higher levels of N in the environment suppress the potential for N-acquiring activity. Nevertheless, further investigation is necessary to test these observations, and more detailed chemical and physical properties of soil should be considered, such as redox potential, which is also a key environmental factor shaping microbial community structure and function (DeAngelis et al. 2010).
Finally, the relationship between soil properties and the relative abundance of various phyla in this study was compared to previous findings. Delta-proteobacteria relative abundance in natural and restored wetland soils was higher than reclaimed soils. The concentrations of TOC was decreased in reclaimed wetland in comparison with that of natural and restored wetland, which supported the previous work that documented Delta-proteobacteria was significantly negatively correlated to organic carbon (Nacke et al. 2011). In contrast, Gamma-proteobacteria relative abundance was higher in reclaimed soils than in natural and restored wetland soils. In this work, reclaimed soils had significantly higher N. Previous work (Campbell et al. 2010) also documented higher abundance of Gamma-proteobacteria in soils with higher nutrient levels.
The relative abundance of Chloroflexi was higher in reclaimed soils than natural soils (Fig. 4). The reclaimed soils in this study were being used to grow rice, and this finding is in agreement with Sheng et al. (2013) who also found a higher relative abundance of Chloroflexi in land. Moreover, it was found that the relative abundance of Chloroflexi was positively correlated with total nitrogen. This result is quite interesting because of its apparent contradiction with previous works, in which Chloroflexi was found to prefer nutrient poor conditions (Ding et al. 2013; Tsitko et al. 2014).These findings illustrated that our understanding of the relative importance of environmental factors influencing Chloroflexi distribution could be increased with research utilizing a range of controlled and manipulated nutrient and oxygen-level conditions. As shown in Fig. 4, Proteobacteria and Acidobacteria (as represented of 49.8% of the bacterial clones) were dominant in all the studied communities which indicated that these phyla may not be largely affected by the changes in land use (Rampelotto et al. 2013). Moreover, quite a large portion of effective bacterial sequences in this study (27.1%) could not be assigned to any taxa at phylum level, indicating the extent of novel sequences captured by this study.
In conclusion, this work demonstrated that microbial community composition and activity were affected by land use types and might be used to assess the extent of recovery of natural microbial-driven processes during wetland restoration. The scope of this work could be extended by utilizing a similar approach to investigate restoration in similar and different types of wetlands, recovering from similar and different types of agriculture, as well as grassland and forest restoration. A collection of studies that provide detailed information would help to facilitate wetland soil management and increase our understanding of the ecological process of natural habitat restoration.
References
Acosta-Martínez V, Dowd S, Sun Y, Allen V (2008) Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biology and Biochemistry 40:2762–2770
Alewell C, Paul S, Lischeid G, Storck FR (2008) Co-regulation of redox processes in freshwater wetlands as a function of organic matter availability? The Science of the Total Environment 404:335–342
Andersen R, Grasset L, Thormann MN, Rochefort L, Francez AJ (2010) Changes in microbial community structure and function following Sphagnum peatland restoration. Soil Biology and Biochemistry 42:291–301
Becker J, Eisenhauer N, Scheu S, Jousset A (2012) Increasing antagonistic interactions cause bacterial communities to collapse at high diversity. Ecology Letters 15:468–474
Bending GD, Turner MK, Jones JE (2002) Interactions between crop residue and soil organic matter quality and the functional diversity of soil microbial communities. Soil Biology and Biochemistry 34:1073–1082
Bisset A, Richardson A, Baker G, Thrall PH (2011) Long-term land use effects on soil microbial community structure and function. Applied Soil Ecology 51:66–78
Blake GR (1965) Bulk density. Methods of soil analysis. Part 1. Physical and mineralogical properties, Including statistics of measurement and sampling, 374–390
Bouyoucos GJ (1962) Hydrometer method improved for making particle size analysis of soils. Agronomy Journal 56:464–465
Campbell BJ, Polson SW, Hanson TE, Mack MC, Schuur EA (2010) The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environmental Microbiology 12:1842–1854
Carbonetto B, Rascovan N, Álvarez R, Mentaberry A, Vázquez MP (2014) Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in Argentine Pampas. PLoS One 9(6):e99949
Castro HF, Classen AT, Austin EE, Norby RJ, Schadt CW (2010) Soil microbial community responses to multiple experimental climate change drivers. Applied and Environmental Microbiology 76:999–1007
Chaudhry V, Rehman A, Mishra A, Chauhan PS, Nautiyal CS (2012) Changes in bacterial community structure of agricultural land due to long-term organic and chemical amendments. Microbial Ecology 64:450–460
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2004) The ribosomal database project (RDP-II): sequences and tools for high- throughput rRNA analysis. Nucleic Acids Research 33:294–296
DeAngelis KM, Silver WL, Thompson AW, Firestone MK (2010) Microbial communities acclimate to recurring changes in soil redox potential status. Environmental Microbiology 12(12):3137–3149
Ding GC, Piceno YM, Heuer H, Weinert N, Dohrmann AB, Carrillo A, Andersen GL, Castellanos T, Tebbe CC, Smalla K (2013) Changes of soil bacterial diversity as a consequence of agricultural land use in a semi-arid ecosystem. PLoS One 8(3):e59497
Doi R, Ranamukhaarachchi SL (2013) Slow restoration of soil microbial functions in an Acacia plantation established on degraded land in Thailand. International Journal of Environmental Science and Technology 10:623–634
Dominati E, Patterson M, Mackay A (2010) A framework for classifying and quantifying the natural capital and ecosystem services of soils. Ecology Economics 69:1858–1868
Drenovsky RE, Steenwerth KL, Jackson LE, Scow KM (2010) Land use and climatic factors structure regional patterns in soil microbial communities. Global Ecology and Biogeography 19:27–39
Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proceedings of the National Academy of Sciences of the United States of America 103:626–631
Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG (2012) Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proceedings of the National Academy of Sciences of the United States of America 109:21390–21395
Fierer N, Ladau J, Clemente JC, Leff JW, Owens SM, Pollard KS, Knight R, Gilbert JA, Gilbert JA (2013) Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342:621–624
Garland JL, Mills AL (1991) Classification and characterization of heterotrophic microbial communities on the basis of patterns of community level sole carbon source utilization. Applied and Environmental Microbiology 57:2351–2359
Girvan MS, Bullimore J, Pretty JN, Osborn AM, Ball AS (2003) Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Applied and Environmental Microbiology 69:1800–1809
Gotelli N, Colwell RK (2001) Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecology Letters 4:379–391
Guan X, Wang J, Zhao H, Wang JJ, Luo X, Liu F, Zhao F (2013) Soil bacterial communities shaped by geochemical factors and land use in a less-explored area, Tibetan Plateau. BMC Genomics 14:820–833
Harris JA (2003) Measurements of the soil microbial community for estimating the success of restoration. European Journal of Soil Science 54:801–808
Hartman WH, Richardson CJ, Vilgaly R, Bruland GL (2008) Environmental and anthropogenic controls over bacterial communities in wetland soils. Proceedings of the National Academy of Sciences of the United States of America 105:17842–17847
Jousset A, Schmid B, Scheu S, Eisenhauer N (2011) Genotypic richness and dissimilarity opposingly affect ecosystem functioning. Ecology Letters 14:537–545
Kubler LA, Miller JO, Ducey TF, Hunt PG, Lang M, Ro KS (2014) Multistate assessment of wetland restoration on CO2 and N2O emissions and soil bacterial communities. Applied Soil Ecology 76:87–94
Legendre P, Legendre L (1998) Numerical Ecology, 2nd English edn, pp. 853. Elsevier, New York
Lewis DE, White JR, Wafula D, Athar R, Dickerson T, Williams HN, Chauhan A (2010) Soil functional diversity analysis of a bauxite-mined restoration chronosequence. Microbial Ecology 59:710–723
Li JH, Jiao SM, Gao RQ, Bardgett RD (2012) Differential effects of legume species on the recovery of soil microbial communities, and carbon and nitrogen contents, in abandoned fields of the loess plateau, China. Environmental Management 50:1193–1203
Lu RK (2000) Methods of soil and agricultural chemistry analysis. Chinese agricultural science and technology Press, Beijing, 1–627
Lu XX, Sun SL, Zhang YQ, Hollibaugh JT, Mou XZ (2015) Temporal and vertical distribution of bacterioplankton at the G ray’s reef national marine sanctuary. Applied and Environmental Microbiology 83(23):910–917
Min Q (1999) Evaluation of the effects of expanding agricultural land use on floods in the Poyang Lake. People Yangtze River 30:30–32
Mitsch WJ, Gosselink JG (2007) Wetlands, fourthed. Wiley, New York
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 6:e17000
Nautiyal CS, Chauhan PS, Bhatia CR (2010) Changes in soil physico-chemical properties and microbial functional diversity due to 14 years of conversion of grassland to organic agriculture in semi-arid agroecosystem. Soil & Tillage Research 109:55–60
Quince C, Lanzén A, Curtis T, Davenport R, Hall N, Head LM, Read LF, Sloan WT (2009) Accurate determination of microbial diversity from 454 pyrosequencing data. Nature Methods 6(9):639–644
Rampelotto PH, Ferrira A, Barboza ADM, Roesch LFW (2013) Changes in diversity, abundance, and structure of soil bacterial communities in Brazilian Savanna under different land use systems. Microbial Ecology 66:593–607
Richardson CJ, Marshall PE (1986) Processes controlling the movement, storage, and export of phosphorus in a fen peatland. Ecology Monographs 56:279–302
Roulet NT (2000) Peatlands, carbon storage, greenhouse gases and the Kyoto protocol: prospects and significance for Canada. Wetlands 20:605–615
Schloss PD, Handelsman J (2004) Status of the microbial census. Microbiology and Molecular Biology Reviews 68:686–691
Schloss PD, Westcott SL, Ryabin T (2009) Introducing mothur: open source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75:7537–7541
Sharma S, Rangger A, von Lützow M, Insam H (1998) Functional diversity of soil bacterial communities increases after maize litter amendment. European Journal of Soil Biology 34:53–60
Sheng R, Meng D, Wu M, Di H, Qin HL, Wei W (2013) Effect of agricultural land use change on community composition of bacteria and ammonia oxidizers. Journal of Soils and Sediments 13:1246–1256
Swift MJ, Heal OW, Anderson JM (1979) Decomposition in terrestrial ecosystems. Blackwell Scientific Publications, Oxford
Tsitko I, Lusa M, Lehto J, Parviainen L, Ikonen AT, Lahdenperä AM, Bomberg M (2014) The variation of microbial communities in a depth profile of an acidic, nutrient-poor boreal bog in southwestern Finland. Open Journal of Ecology 4:832–859
Turner S, Pryer KM, Miao VP, Palmer JD (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. Journal of Eukaryotic Microbiology 46:327–338
US Environmental Protection Agency (2014) Soil sampling operation procedures
van Dijk J, Didden WA, Kuenen F, van Bodegom PM, Verhoef HA, Aerts R (2009) Can differences in soil community composition after peat meadow restoration lead to different decomposition and mineralization rates? Soil Biology and Biochemistry 41:1717–1725
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73:5261–5267
World Conservation Union (1996) Guidelines for aid agencies for improved conservation and sustainable use of tropical and sub-tropical wetlands. Organization for Economic Cooperation and Development, Paris
Yu WT, Bi ML, Xu YG, Zhou H, Ma Q, Jiang CM (2013) Microbial biomass and community composition in a Luvisol soil as influenced by long-term land use and fertilization. Catena 107:89–95
Zak DR, Holmes WE, White DC, Peacock AD, Tilman D (2003) Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84:2042–2050
Zhang T, Shao MF, Ye L (2012) 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. The ISME Journal 6:1137–1147
Zhao D, Li F, Yang Q, Wang R, Song Y, Tao Y (2013) The influence of different types of urban land use on soil microbial biomass and functional diversity in Beijing, China. Soil Use and Management 29:230–239
Acknowledgements
This research was supported by the National Natural Science Foundations of China (Grant No. 31360127, 30860062 and 31660149) and the Open Foundation of MOE Key Laboratory of Poyang Lake Environment and Resource (PYH2015-13) and Nanchang University Seed Grant for Biomedicine (9202-0210210807). We appreciate Dr. Rong J and Lan ZC for invaluable comments on early drafts of this paper, and we also thank two anonymous reviewers for providing comments that helped us improve this paper.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Huang, L.L., Kou, W.B., Wu, L. et al. Microbial Composition and Activity of Natural, Restored, and Reclaimed Wetland Soils: a Case Study of Poyang Lake Basin, China. Wetlands 39 (Suppl 1), 113–123 (2019). https://doi.org/10.1007/s13157-018-1020-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13157-018-1020-y