
Abstract
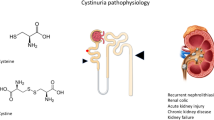
Cystinuria, a genetic disorder of cystine transport, is characterized by excessive excretion of cystine in the urine and recurrent cystine stones in the kidneys and, to a lesser extent, in the bladder. Males generally are more severely affected than females. The disorder may lead to chronic kidney disease in many patients. The cystine transporter (b0,+) is a heterodimer consisting of the rBAT (encoded by SLC3A1) and b0,+AT (encoded by SLC7A9) subunits joined by a disulfide bridge. The molecular basis of cystinuria is known in great detail, and this information is now being used to define genotype–phenotype correlations. Current treatments for cystinuria include increased fluid intake to increase cystine solubility and the administration of thiol drugs for more severe cases. These drugs, however, have poor patient compliance due to adverse effects. Thus, there is a need to reduce or eliminate the risks associated with therapy for cystinuria. Four mouse models for cystinuria have been described and these models provide a resource for evaluating the safety and efficacy of new therapies for cystinuria. We are evaluating a new approach for the treatment of cystine stones based on the inhibition of cystine crystal growth by cystine analogs. Our ongoing studies indicate that cystine diamides are effective in preventing cystine stone formation in the Slc3a1 knockout mouse model for cystinuria. In addition to crystal growth, crystal aggregation is required for stone formation. Male and female mice with cystinuria have comparable levels of crystalluria, but very few female mice form stones. The identification of factors that inhibit cystine crystal aggregation in female mice may provide insight into the gender difference in disease severity in patients with cystinuria.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In this review, we summarize established knowledge on cystinuria; provide a perspective on how patients view their disease; emphasize the limitations of current treatment modalities; outline progress in understanding the relationship between genotype and phenotype; describe what we have learned from mouse models for cystinuria; present a new approach for therapy based on inhibition of cystine crystal growth; and, finally, highlight cystine crystal aggregation as a major area for future investigation.
Cystinuria
Transport defect
Cystinuria (OMIM #220100) is a genetic disorder in the transport of cystine and the dibasic amino acids ornithine, lysine and arginine (COLA) in the renal proximal tubules [1,2,3,4,5,6,7]. Of the four amino acids, cystine is the least soluble and it can crystallize in the urinary tract, leading to cystine stone formation. The COLA transporter (b0,+) is a heterodimer consisting of the rBAT (encoded by SLC3A1, located on chromosome 2p16.3) and b0,+AT (encoded by SLC7A9, located on chromosome 19q13.1) subunits joined by a disulfide bridge.
Cystinuria is a chronic disorder characterized by the hyper-excretion of cystine in the urine (> 400 mg/day compared with < 30 mg/day in normal subjects) and development of cystine stones in the kidney and, to a lesser extent, in the bladder [8,9,10]. It accounts for 1–2% and 6–8% of urinary tract stones in adults and children, respectively [11, 12], making it the most common genetic cause of urolithiasis in the pediatric population. The same defect is present in the small intestine, but this is considered to be of little consequence as amino acids are primarily absorbed as small peptides [13, 14].
Prevalence
The prevalence of cystinuria globally is estimated to be 1 per 7000 with widespread variation, ranging from 1 in 2500 in Israeli Jews of Libyan origin (due to a founder effect) to 1 in 100,000 in Sweden [2, 3, 8]. The estimated prevalence in the US population is 1 in 10,000, suggesting that there are approximately 33,000 affected individuals [15]. However, the true prevalence may be higher as some affected individuals do not form stones or form them so infrequently that the diagnosis of cystinuria is not made [8].
Clinical presentation
Although stones may form at any age, stone formation in cystinuria most commonly occurs within the first two decades of life, with approximately 50% of patients developing their first stone in the first decade of life and 25–40% during their teenage years [2, 8, 16, 17]. About 75% of these patients will present with bilateral stones [8].The diagnosis is usually made by confirming the stone composition as cystine. Siblings of patients with cystinuria should be screened by measurement of urinary cystine by adding nitroprusside to the urine, which turns reddish/purple in the presence of excess cystine.
Males are generally more severely affected than females and they have a larger number of stones [2]. On average, untreated patients experience one new stone every year and undergo a surgical procedure to remove the stones every 3 years. By middle age, the average cystinuria patient will typically undergo seven surgical procedures [18]. In a longitudinal study involving 16 patients, it was shown that patients who received early medical management after the initial presentation had the lowest recurrence rates and they tended to maintain their renal function, emphasizing the importance of prompt metabolic assessment [19].
Kidney disease
Up to 70% of patients with cystinuria may develop chronic kidney disease (CKD), which may lead to end-stage renal disease (ESRD) [16, 18]. In a retrospective study that collected data from 442 patients with cystinuria, 27% of patients had an estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2, indicating some degree of CKD. Hypertension was also common in this study, with 28.6% of patients having high blood pressure [20]. Bilateral stones may also increase the risk for acute kidney injury, particularly in children. It has been suggested that young children with acute renal failure due to ureteral stones should be tested for cystinuria [21]. Compared with calcium oxalate stone formers, patients with cystinuria are more likely to have abnormal serum creatinine levels and they are at higher risk for nephrectomy [15, 22].
Cystinuria genotypes
Cystinuria genotypes are classified as Type A or Type B. Type A is caused by mutations in SLC3A1 and Type B by mutations in SLC7A9. Clinical symptoms in the A and B sub-groups are similar and there is no apparent correlation between genotype and phenotype but, as noted below, relationships between genotypes and phenotypes are now beginning to be delineated. SLC3A1 heterozygotes have no apparent phenotype, but SLC7A9 heterozygotes excrete variable levels of COLA in the urine [2, 23]. Few of these latter patients will have stones, but perhaps they are more likely to do so if urine volumes are low or animal-protein ingestion is high.
The percentage of Type A and B genotypes appears to be population-dependent, but the distribution may be skewed by the inevitably small number of patient samples in the various studies. There is a preponderance of mutations in SLC7A9 in the Spanish population, but a higher percentage (55–75%) of mutations in SLC3A1 has been reported in other European populations (UK, France, Eastern Europe) and an equal distribution has been found in the American population. Approximately 2% of patients have mutations in at least one copy of each gene, but stone formation only occurs if both copies of either gene are abnormal (Type AAB or ABB cystinuria). Mutations have not been identified or only one mutant allele has been identified in approximately 5% of patients with cystinuria [5, 16, 17, 24, 25]. To date, 241 mutations have been identified in SLC3A1 and 159 mutations in SLC7A9, including atypical cases for both genes, in the Human Gene Mutation Database (HGMD® Professional 2018, http://www.hgmd.cf.ac.uk/ac/index.php, accessed November 12, 2018) [26].
Limitations of current treatment modalities
Medical treatment for cystinuria involves increasing cystine solubility by increasing urine volume and pH. Hyperdiuresis is the most important factor for preventing cystine stone formation, but this requires water intake of > 4 liters/day and urine output of > 3 liters/day [2, 6, 23], which is difficult to achieve in young children. Cystine solubility increases with pH but only at values greater than 7.0, which is difficult to maintain on a long-term basis and may predispose to formation of calcium phosphate stones [27]. The use of thiols d-penicillamine (Cuprimine®) and tiopronin (Thiola®) are options for the most seriously affected patients [6, 28]. These sulfhydryl drugs convert cystine to the more soluble mixed disulfides of cysteine-penicillamine or cysteine-tiopronin, respectively. Both drugs require the use of high doses (1–2 g/day) and are associated with allergy and toxicity, limiting their use.
Despite preventative measures, most patients with cystinuria eventually require multiple surgical procedures for stone management. Surgical options include extracorporeal shockwave lithotripsy (ESWL), but cystine stones are somewhat resistant to ESWL and may require multiple rounds of treatment with attendant risk of kidney injury [29]. Patient compliance with dietary therapy is often poor; sulfhydryl drugs have multiple side effects; and repeated surgical interventions can lead to renal insufficiency. Therefore, current therapies for cystinuria are inadequate.
Clinical trials
The thiol drugs d-penicillamine and tiopronin are part of the standard treatment regimen for cystinuria, but they are associated with significant side effects, including allergic reactions, skin disorders, liver abnormalities, nephrotic syndrome, and blood disorders [30]. Clinical trials are currently in progress aimed at reducing the side effects associated with cystinuria therapy (http://www.clinicaltrials.gov, accessed November 12, 2018). These include bucillamine, a thiol-binding drug developed from tiopronin (NCT02942420); tolvaptan, a vasopressin antagonist that increases urinary output (NCT02538016); and α-lipoic acid, a nutritional supplement that has been shown to increase cystine solubility in the Slc3a1 knockout mouse model for cystinuria (NCT02910531) [31]. Two recently completed clinical trials involved measurement of cystine supersaturation as a predictor of cystine stone recurrence (NCT02120105) and the relationship between dosage of cystine-binding thiol drugs and cystine concentration in the urine (NCT02125721). An earlier trial involving a mixture of nine Asian Indian herbs collectively known as Cystone did not decrease urine cystine levels or stone burden in patients with cystinuria (NCT00381849) [32].
Quality of life issues
A number of surveys have evaluated the health-related quality of life (HRQoL) issues in patients who form cystine stones compared with non-cystine stone formers or the general public [33,34,35]. Using the Rand Medicine Survey Form 36 (SF-36), a generic form for self-reporting of quality of life measures, Modersitzki et al. [33] noted that, in a sample of 214 cystine stone formers and 81 non-cystine stone formers, the former group suffered longer from kidney stones and had significantly more episodes of stones than the latter group and that all stone formers, irrespective of stone type, had worse HRQoL scores than the general US population. In the Parr et al. SF-36 survey [34], based on a sample size of 14 patients, cystinuria was associated with a higher rate of surgical intervention and a lower QoL compared with the general population. Further, patients reported that medical management was either ineffective or poorly tolerated, at least in part due to the adverse side effects of available medications. In the Streeper et al. study [35], using the disease-specific Wisconsin Stone Quality of Life (WISQOL) questionnaire in a sample of 12 cystine stone formers and 90 patients with mixed calcium stones, patients with cystine stones had lower WISQOL scores and lower HRQoL scores compared with non-cystine stone formers. Collectively, these studies highlight the importance of improving the medical management of patients with cystinuria to reduce the rate of morbidity and surgical interventions.
Associated disorders
Cystinuria normally manifests as a renal system disorder, but several patients have been reported in which cystinuria is associated with neurological, muscular, mitochondrial, or other abnormalities. These include 2p21 deletion syndrome, hypotonia-cystinuria syndrome (HCS), and atypical HCS [36,37,38]. The 2p21 deletion syndrome is characterized by moderate–severe psychomotor retardation and a decrease in activity of the respiratory chain complexes I, III, IV and V. Patients with HCS have generalized hypotonia, growth retardation and minor facial dysmorphic features. The phenotype in atypical HCS is similar to that in classical HCS but with mild–moderate mental retardation and a decrease in activity of only respiratory chain complex IV. Genomic alterations in these patients include contiguous gene deletions involving SLC3A1 and one or more other genes, with the severity of the different syndromes reflecting the number of genes that are deleted. The deletion spans SLC3A, PREPL, C2orf34 and PPM1B in 2p21 deletion syndrome; SLC3A1, PREPL and C2orf34 in atypical HCS; and SLC3A1 and PREPL in HCS.
A contiguous gene deletion involving SLC7A9 has recently been described in a patient with the 19q13.1 microdeletion syndrome [39]. Clinical features of this syndrome include facial dysmorphism, ectodermal dysplasia, growth retardation, neurologic features and genitourinary anomalies. The patient had elevated levels of cystine and lysine in the urine but no urinary tract stones. A 5.81 Mb deletion spanning 19 genes, including SLC7A9, was identified in one allele and the other SLC7A9 allele had a point mutation. SLC7A9 deletions have been suggested, but not verified, in several patients with elevated COLA levels in the urine but with an identified mutation in only one allele of SLC7A9. A Korean boy with mental retardation, ataxia and hyper-excretion of COLA but no renal stones had a heterozygous mutation (G173R) in SLC7A9. The same mutation was present in his mother and sister, but extensive analysis of SLC7A9 and SLC3A1 did not identify the second mutation in this patient. DNA from the father was not available for testing. The second mutation could be a contiguous gene deletion spanning SLC7A9 inherited from the father [40].
A Japanese patient with elevated excretion of COLA but no urinary stones and with atrophy in the cerebellum, brainstem and cerebrum had a heterozygous mutation (P482L) in SLC7A9, again suggesting the possibility that the second mutation is a large deletion [41]. A homozygous point mutation (F4976L) was identified in the ryanodine receptor type 1 (RYR1) gene in two sisters from a consanguineous family with cystinuria due to a point mutation in SLC7A9 and congenital myopathy. RYR1 is located telomeric to SLC7A9 in a 5 Mb region on chromosome 19q13.1, thus explaining the association between these two disorders. No deletions or insertions were identified in the sequenced region, suggesting that the co-occurrence of these two conditions is due to the inheritance of the same haplotype from both parents [42].
Recent findings
Genotype–phenotype correlations
Analysis of genetic alterations and their functional effects at the protein level may allow prediction of the clinical course of a disease as well as identification of modifying genetic, epigenetic or environmental factors that influence clinical outcome. This approach has been demonstrated for diseases such as Lesch–Nyhan disease [43] and ATP-binding cassette transporter A3 (ABCA3) deficiency [44]. As indicated earlier, 241 mutations in SLC3A1 and 159 mutations in SLC7A9 are listed in the Human Gene Mutation Database. Observational studies have generally not identified specific genotype–phenotype correlations for cystinuria [16, 45], but preliminary analysis of genetic data from 74 patients showed that the presence of at least one missense mutation in SLC3A1 correlated with lower urinary levels of ornithine, lysine and arginine, but not of cystine, compared with all other combinations of mutations [17].
The relationship between cystinuria genotypes and phenotypes is now being explored in more detail using a variety of computational methods [46,47,48]. As an example, computer modeling of b0+AT (encoded by SLC7A9), suggested that mutations such as p.Arg171Trp and p.Arg333Trp are located close to the end of the transport membrane channel and they could affect membrane stability [48]. Computational predictions can: (i) provide better understanding of genotype–phenotype relationships; (ii) facilitate the development of new diagnostic markers; and (iii) eventually lead to personalized treatment options [46, 47]. Ultrasound scanning during the 22–33 weeks of gestation has identified hyperechoic colon (HEC) in some fetuses with cystinuria. DNA sequence analysis from newborns with or without HEC indicated that only patients with more severe mutations within SLC3A1 or SLC7A9 presented with HEC, suggesting another relationship between genotype and phenotype [49].
SLC7A13 and isolated cystinuria
Early studies on lysine and arginine infusion in control subjects and in patients with cystinuria suggested the presence of two transport systems for cystine in the kidney: a low capacity system that is defective in patients with cystinuria and a high capacity system that is intact in these patients [50]. Three additional lines of evidence support the presence of a second transporter for cystine. First, atypical cystinuria has been described in a British family and in a German family in which probands had hypercystinuria without a concomitant increase in the excretion of ornithine, lysine and arginine [51, 52]. Second, the primary COLA transporter (b0,+) is located in the S1 segment of proximal renal tubules, but the expression of rBAT is most abundant and that of b0,+AT least abundant in the S3 segment, suggesting the presence of another partner for rBAT in this segment [53, 54]. Third, in Slc7a9 knockout mice [55], the fractional excretion of cystine is 11%, suggesting the reabsorption of cystine through an uncharacterized transporter [56]. The nature of this transporter has remained elusive until recently. AGT1 (encoded by SLC7A13) has now been identified as the partner of rBAT in the S3 segment [57]. It remains to be determined whether mutations in SLC7A13 are the cause of isolated cystinuria.
Mouse models
Knockout and point-mutation models
Four mouse models for cystinuria have been described to date. These are Slc3a1 knockout (exon 1 deletion), induced mutation (D140G), and spontaneous mutation (E383K), respectively, for Type A and Slc7a9 knockout (exons 3–9 deletion) for Type B (Table 1) [31, 55, 58,59,60]. A model for Type AB cystinuria (Slc3a1 and Slc7a9 double heterozygous mice, Slc3a1+/−Slc7a9+/−) also has been generated by crossing Slc3a1 mice with the induced mutation and Slc7a9 knockout mice (Table 1) [61]. We recently compared the biochemical and histopathological findings in our Slc3a1 knockout mice with the other three mouse models (Sahota et al. submitted). Our findings are summarized below.
Cystine stone formation in mutant mice was evident within the first two months of life replicating, to a large extent, the corresponding human cystinuria phenotypes. Mice with two mutant copies of Slc3a1 (irrespective how they were generated) excreted significantly higher levels of COLA compared with their wild-type counterparts, but amino acid excretion in heterozygous mice was indistinguishable from that in wild-type mice [60]. Slc7a9 knockout mice also showed elevated excretion of COLA and Slc7a9 heterozygotes showed moderate but significant excretion of these amino acids [55]. As indicated earlier, a small number of patients with the rare type AB cystinuria have been reported, but these individuals actually have two mutant copies of one gene and a mutant copy of the other gene (genotype AAB or ABB) [6, 17, 24]. Four percent of double heterozygous mice showed cystine stone formation, suggesting possible digenic inheritance [61].
Gender differences in disease severity
Males with cystinuria are affected more frequently and more severely, and they have an earlier onset of stone formation and a larger number of stones compared with females [3]. This phenotypic characteristic is also evident in Slc3a1 mouse models of cystinuria. Microcrystalluria was present in female mice with the induced mutation in Slc3a1, but these mice showed a delayed onset or reduced penetrance of stone formation compared with males [60]. In the Slc3a1 mouse model with the spontaneous mutation, cystine crystals were present in the urine from both males and females but urolithiasis affected mainly males [59]. In Slc3a1 knockout mice, crystal number and size distribution of crystals in urine samples from females were similar to those in males, but bladder stones were detected in only a few female mice and then only after age 18 months (Sahota et al. submitted).
In contrast, sex differences in stone formation were not noted in Slc7a9 knockout mice [55]. These mice formed stones from age 1 month in an approximate 1:1 male/female ratio and this ratio was maintained throughout the lifespan of the animals. The basis for the increased disease severity in Slc7a9 knockout females compared with Slc3a1 knockout females is not known, but it may be due to differences in cystine crystal aggregation between different mouse strains (see Future direction).
Genetic and environmental factors
The wide phenotypic variability in patients with cystinuria, even in families with the same mutation, suggests that modifying genes or environmental factors contribute to the phenotype [62]. These influences also are evident in mouse models for cystinuria. We have shown that Slc3a1 knockout male and female mice have crystalluria but only about 60% of male mice form stones by 16 weeks of age and only a few female mice form stones by 18 months (Sahota et al. submitted). In another study [55], approximately 82% of Slc7a9 knockout mice had crystalluria and 42% had cystine stones at age 1 month, but approximately 2% were stone-free up to age 1 year. These findings suggest that modifier genes or epigenetic effects influence the cystinuria phenotype but no such effect has been identified to date. The contribution of diet to the propensity for stone formation in the various mouse models also has not been fully evaluated.
COLA deficits in plasma
Patients with cystinuria generally have lower plasma levels of COLA due to the excessive excretion of these amino acids in the urine, but this loss can be compensated to a large extent through the absorption of oligopeptides [13, 14]. Lower plasma levels of COLA also have been observed in mouse models of cystinuria, but this is gender- and strain-dependent. We observed lower levels of COLA and lower levels of OLA in Slc3a1 knockout male and female mice, respectively, compared with their wild-type counterparts (Sahota et al. submitted). Slc7a9 knockout mice exhibited a 30% decrease in plasma cystine and lysine levels, but no differences were observed for the other dibasic amino acids [55]. In Slc3a1 mice with the induced or spontaneous mutation, on the other hand, plasma levels of OLA did not differ among genotypes, indicating that the mutant animals can compensate completely for the urinary loss of these amino acids [60].
Kidney disease
CKD in patients with cystinuria may result from recurrent stones, repeated urologic interventions, or obstructive uropathy [2, 63]. In mouse models of cystinuria, cystine stone formation occurs primarily in the bladder but there are age- and strain-dependent changes in renal histopathology that likely are due to obstructive uropathy. At age 3 months, plasma levels of creatinine and blood urea nitrogen in Slc3a1 knockout males were similar to those in wild-type males (Sahota et al. submitted). In addition, there were no changes in renal pathology at this age but progressive changes, including tubular atrophy and tubulointerstitial nephritis, were observed at age 5 months and beyond. At age 4 months, Slc3a1 mice with the spontaneous mutation had cystine casts in the tubules that were associated with focal inflammatory cell infiltrates in the cortex [59]. In Slc3a1 mice with the induced mutation, cystine lithiasis led to cystic pelvis dilatation and atrophy of renal cortex and medulla at age 3 months [60]. Kidneys from stone-bearing Slc7a9 knockout mice 4 months of age or older showed severe tubular and pelvic dilatation, tubular necrosis and chronic interstitial nephritis, or tubular hyaline droplets [55].
Primary site of cystine stone formation
As outlined above, there are multiple similarities between human and mouse cystinuria but the initial site of stone formation in the two species is different. In patients with cystinuria, stones are seen predominantly in the kidney and ureter [64] and instances of cystine stones in the bladder are rare [65, 66]. Mice, on the other hand, initially form cystine stones in the bladder but, with advancing bladder obstruction in older male mice, stones are observed in the upper urinary tract as well [55, 59, 60]. The bladder also is the major site of cystine stone formation in other animals with cystinuria [67]. In animal models, including Slc3a1 mutant mice, the prevalence and/or severity of bladder stone disease in females is lower than that in males and this has been ascribed to the shorter urethra in females [68]. The observation that Slc7a9-/- male and female mice have approximately equal prevalence of bladder stones [55] suggests that components in the urine that promote or inhibit crystal aggregation may also be contributing factors to the propensity for stone formation (see Future direction).
Evaluation of new therapies
The availability of mouse models provides a resource for evaluating the safety and efficacy of new therapies for cystinuria. Font-Llitjo´s et al. [69] described a protocol to assess the therapeutic effects of the thiol drug D-penicillamine in the Slc7a9 knockout mouse model. As described below, we are evaluating a new approach for the treatment of cystinuria based on the inhibition of cystine crystal growth by cystine analogs and we have demonstrated the utility of this approach in the Slc3a1 knockout mouse model [70,71,72,73]. We have also demonstrated that α-lipoic acid inhibits cystine stone growth in this model [31].
Inhibition of cystine crystal growth as a therapy for cystinuria
Cystine stones are difficult to manage medically or surgically and drug therapy in severe cases is limited to two thiol drugs that have poor patient compliance due to adverse effects [6, 28]. The high recurrence rate and the need for multiple treatments can lead to progressive decline in renal function [2, 20, 74]. Thus, there is a need to reduce or eliminate the risks associated with therapy for cystinuria.
Ward and colleagues described a strategy that interferes with the earliest stages in the stone formation process, i.e., inhibition of crystal growth [71, 75,76,77]. Using real-time in situ atomic force microscopy (AFM) and bulk crystallization studies, they demonstrated that low concentrations of cystine mimics, such as cystine dimethyl ester (CDME), bind to preferred sites on cystine crystal surfaces, thus retarding further growth of these crystals in vitro [76]. This was accompanied by a dramatic reduction in crystal size as well as a change in crystal habit. This strategy offers an innovative approach for the treatment of cystinuria.
As a proof of principle, we assessed the effectiveness of CDME as an inhibitor of cystine crystal growth for the treatment of cystine urolithiasis in the Slc3a1 knockout mouse model of cystinuria [73]. Our studies demonstrated that in the presence of CDME the stone burden was decreased by 50% and the stones formed were smaller in size and more numerous compared with stones from untreated mice. This is consistent with the hypothesis that CDME inhibits cystine crystal growth. There are, however, limitations to using CDME as the basis for further drug development because this compound is an ester and it would be expected to be hydrolyzed to cystine by intestinal and plasma esterases, thus limiting its bioavailability. Hu and colleagues designed and synthesized a series of cystine diamides with greater stability and bioavailability compared with CDME. One of the most effective inhibitors synthesized to date is cystine bis(Nʹ-methylpiperazide), denoted LH708, which has been shown to be effective in inhibiting cystine stone formation in Slc3a1 knockout mice [70, 72, 78]. Our studies, to date, strongly support the evaluation of this novel chemical entity and its analogs as a potential therapy for cystinuria.
Future direction
The presence of cystine crystals in the urine is a necessary but not a sufficient condition for the formation of cystine stones. Female patients with cystinuria have slightly higher concentration of cystine in the urine than males, but they exhibit a lower frequency of renal stone occurrence [79]. Thus, the severity of the clinical phenotype among patients cannot be explained by differences in urine cystine excretion alone [80]. Masotti et al. reported that, based on LCMS analysis of normal urine samples in the presence of increasing levels of exogenously added cystine, the solubility of cystine was markedly higher in female samples than in male samples [81]. This inversely correlated with the levels of purine nucleosides (adenosine, guanosine, and inosine) and vanillylmandelic acid (VMA) in the urine, with lower levels of these constituents favoring cystine solubility. Slc3a1 knockout female mice have cystine crystals in urine at levels similar to those in knockout male mice, but only a few females form cystine stones by age 18 months. Slc3a1 knockout male mice, on the other hand, form cystine stones from age 6–8 weeks (Sahota et al. submitted).
In addition to crystal nucleation and growth, crystal aggregation is required for stone formation [82,83,84,85], but the factors that regulate cystine crystal aggregation are not known. The role of crystal aggregation has been well established for calcium oxalate stones [85,86,87], suggesting that aggregation is likely to be important for cystine stone formation as well. Metabolomic and proteomic analyses of urine samples from patients with cystinuria and from different mouse models of cystinuria may identify components in the urine that promote or inhibit cystine crystal aggregation. These findings may lead to a better understanding of the gender differences in disease severity in patients with cystinuria.
References
Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJ (2016) How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis 44:65–76. https://doi.org/10.1007/s00240-015-0841-x
Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R (2013) Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol 28:1923–1942. https://doi.org/10.1007/s00467-012-2329-z
Eggermann T, Venghaus A, Zerres K (2012) Cystinuria: an inborn cause of urolithiasis. Orphanet J Rare Dis 7:19. https://doi.org/10.1186/1750-1172-7-19
Pereira DJ, Schoolwerth AC, Pais VM (2015) Cystinuria: current concepts and future directions. Clin Nephrol 83:138–146
Saravakos P, Kokkinou V, Giannatos E (2014) Cystinuria: current diagnosis and management. Urology 83:693–699. https://doi.org/10.1016/j.urology.2013.10.013
Sumorok N, Goldfarb DS (2013) Update on cystinuria. Curr Opin Nephrol Hypertens 22:427–431. https://doi.org/10.1097/MNH.0b013e3283621c5d
Thomas K, Wong K, Withington J, Bultitude M, Doherty A (2014) Cystinuria-a urologist’s perspective. Nat Rev Urol 11:270–277. https://doi.org/10.1038/nrurol.2014.51
Biyani CS, Cartledge JJ (2006) Cystinuria—diagnosis and management. EAU-EBU Update Ser 4:175–183
Schwartz BF, Stoller ML (2000) The vesical calculus. Urol Clin N A 27:333–346
Schwentner C, Oswald J, Lunacek A, Bartsch G, Radmayr C (2005) Giant cystine stone in an infant bladder with no evidence of cystinuria-valence of possible pathomechanisms. Urol Int 75:285–287. https://doi.org/10.1159/000087810
Ahmed K, Dasgupta P, Khan MS (2006) Cystine calculi: challenging group of stones. Postgrad Med J 82:799–801. https://doi.org/10.1136/pgmj.2005.044156
Knoll T, Zollner A, Wendt-Nordahl G, Michel MS, Alken P (2005) Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol 20:19–24. https://doi.org/10.1007/s00467-004-1663-1
Asatoor AM, Freedman PS, Gabriel JR, Milne MD, Prosser DI, Roberts JT, Willoughby CP (1974) Amino acid imbalance in cystinuria. J Clin Pathol 27:500–504
Hellier MD, Perrett D, Holdsworth CD (1970) Dipeptide absorption in cystinuria. Br Med J 4:782–783
Goldstein B, Goldfarb DS (2017) Early recognition and management of rare kidney stone disorders. Urol Nurs 37:81–89, 102
Rhodes HL, Yarram-Smith L, Rice SJ, Tabaksert A, Edwards N, Hartley A, Woodward MN, Smithson SL, Tomson C, Welsh GI, Williams M, Thwaites DT, Sayer JA, Coward RJ (2015) Clinical and genetic analysis of patients with cystinuria in the United Kingdom. CJASN 10:1235–1245. https://doi.org/10.2215/cjn.10981114
Wong KA, Mein R, Wass M, Flinter F, Pardy C, Bultitude M, Thomas K (2015) The genetic diversity of cystinuria in a UK population of patients. BJU Int 116:109–116. https://doi.org/10.1111/bju.12894
Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P (2000) Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 163:1419–1423
Moore SL, Somani BK, Cook P (2018) Journey of a cystinuric patient with a long-term follow-up from a medical stone clinic: necessity to be SaFER (stone and fragments entirely removed). Urolithiasis. https://doi.org/10.1007/s00240-018-1059-5
Prot-Bertoye C, Lebbah S, Daudon M, Tostivint I, Bataille P, Bridoux F, Brignon P, Choquenet C, Cochat P, Combe C, Conort P, Decramer S, Dore B, Dussol B, Essig M, Gaunez N, Joly D, Le Toquin-Bernard S, Mejean A, Meria P, Morin D, N’Guyen HV, Noel C, Normand M, Pietak M, Ronco P, Saussine C, Tsimaratos M, Friedlander G, Traxer O, Knebelmann B, Courbebaisse M (2015) CKD and Its risk factors among patients with cystinuria. CJASN 10:842–851. https://doi.org/10.2215/cjn.06680714
Nalcacioglu H, Ozden E, Genc G, Yakupoglu YK, Sarikaya S, Ozkaya O (2013) An uncommon cause of acute kidney injury in young children: cystinuria. J Pediatr Urol 9:e58–e63. https://doi.org/10.1016/j.jpurol.2012.08.006
Assimos DG, Leslie SW, Ng C, Streem SB, Hart LJ (2002) The impact of cystinuria on renal function. J Urol 168:27–30
Goldfarb DS (2011) Potential pharmacologic treatments for cystinuria and for calcium stones associated with hyperuricosuria. CJASN 6:2093–2097. https://doi.org/10.2215/cjn.00320111
Font-Llitjos M, Jimenez-Vidal M, Bisceglia L, Di Perna M, de Sanctis L, Rousaud F, Zelante L, Palacin M, Nunes V (2005) New insights into cystinuria: 40 new mutations, genotype–phenotype correlation, and digenic inheritance causing partial phenotype. J Med Genet 42:58–68. https://doi.org/10.1136/jmg.2004.022244
Gaildrat P, Lebbah S, Tebani A, Sudrie-Arnaud B, Tostivint I, Bollee G, Tubeuf H, Charles T, Bertholet-Thomas A, Goldenberg A, Barbey F, Martins A, Saugier-Veber P, Frebourg T, Knebelmann B, Bekri S (2017) Clinical and molecular characterization of cystinuria in a French cohort: relevance of assessing large-scale rearrangements and splicing variants. Mol Genet Genom Med 5:373–389. https://doi.org/10.1002/mgg3.294
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN (2017) The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 136:665–677. https://doi.org/10.1007/s00439-017-1779-6
Johri N, Cooper B, Robertson W, Choong S, Rickards D, Unwin R (2010) An update and practical guide to renal stone management. Nephron Clin Pract 116:c159–171. https://doi.org/10.1159/000317196
DeBerardinis RJ, Coughlin CRII, Kaplan P (2008) Penicillamine therapy for pediatric cystinuria: experience from a cohort of American children. J Urol 180:2620–2623. https://doi.org/10.1016/j.juro.2008.08.057
Wood K, Keys T, Mufarrij P, Assimos DG (2011) Impact of stone removal on renal function: a review. Rev Urol 13:73–89
Asplin JR, Penniston K, Goldfarb DS (2013) Monosodium urate stones are rare, and urine pH is not low in cystinuria. Am J Kidney Dis 62:179–180. https://doi.org/10.1053/j.ajkd.2013.03.041
Zee T, Bose N, Zee J, Beck JN, Yang S, Parihar J, Yang M, Damodar S, Hall D, O’Leary MN, Ramanathan A, Gerona RR, Killilea DW, Chi T, Tischfield J, Sahota A, Kahn A, Stoller ML, Kapahi P (2017) α-Lipoic acid treatment prevents cystine urolithiasis in a mouse model of cystinuria. Nat Med 23:288–290. https://doi.org/10.1038/nm.4280
Erickson SB, Vrtiska TJ, Canzanello VJ, Lieske JC (2011) Cystone(R) for 1 year did not change urine chemistry or decrease stone burden in cystine stone formers. Urol Res 39:197–203. https://doi.org/10.1007/s00240-010-0334-x
Modersitzki F, Pizzi L, Grasso M, Goldfarb DS (2014) Health-related quality of life (HRQoL) in cystine compared with non-cystine stone formers. Urolithiasis 42:53–60. https://doi.org/10.1007/s00240-013-0621-4
Parr JM, Desai D, Winkle D (2015) Natural history and quality of life in patients with cystine urolithiasis: a single centre study. BJU Int 116(Suppl 3):31–35. https://doi.org/10.1111/bju.13169
Streeper NM, Wertheim ML, Nakada SY, Penniston KL (2017) Cystine stone formers have impaired health-related quality of life compared with noncystine stone formers: a case-referent study piloting the Wisconsin stone quality of life questionnaire among patients with cystine stones. J Endourology 31(S1):S48–Ss53. https://doi.org/10.1089/end.2016.0564
Chabrol B, Martens K, Meulemans S, Cano A, Jaeken J, Matthijs G, Creemers JW (2009) Deletion of C2orf34, PREPL and SLC3A1 causes atypical hypotonia-cystinuria syndrome. BMJ Case Rep. https://doi.org/10.1136/bcr.08.2008.0719
Martens K, Jaeken J, Matthijs G, Creemers JW (2008) Multi-system disorder syndromes associated with cystinuria type I. Curr Mol Med 8:544–550
Parvari R, Brodyansky I, Elpeleg O, Moses S, Landau D, Hershkovitz E (2001) A recessive contiguous gene deletion of chromosome 2p16 associated with cystinuria and a mitochondrial disease. Am J Hum Genet 69:869–875. https://doi.org/10.1086/323624
de Rojas T, Aparicio C, de Lucas C, Martinez B, Gil-Fournier B, Ramiro-Leon S (2016) Cystinuria in a patient with 19q12q13.1 deletion. CEN Case Rep 5:67–69. https://doi.org/10.1007/s13730-015-0193-y
Lee EH, Kim YH, Hwang JS, Kim SH (2010) Non-type I cystinuria associated with mental retardation and ataxia in a Korean boy with a new missense mutation (G173R) in the SLC7A9 gene. J Korean Med Sci 25:172–175. https://doi.org/10.3346/jkms.2010.25.1.172
Tohge R, Sakamoto S, Takahashi M (2016) A case of cystinuria presenting with cerebellar ataxia and dementia. Pract Neurol 16:296–299. https://doi.org/10.1136/practneurol-2016-001374
Astrea G, Munteanu I, Cassandrini D, Lillis S, Trovato R, Pegoraro E, Cioni G, Mercuri E, Muntoni F, Battini R (2015) A diagnostic dilemma in a family with cystinuria type B resolved by muscle magnetic resonance. Pediatr Neurol 52:548–551. https://doi.org/10.1016/j.pediatrneurol.2015.01.018
Fu R, Ceballos-Picot I, Torres RJ, Larovere LE, Yamada Y, Nguyen KV, Hegde M, Visser JE, Schretlen DJ, Nyhan WL, Puig JG, O’Neill PJ, Jinnah HA (2014) Genotype–phenotype correlations in neurogenetics: Lesch–Nyhan disease as a model disorder. Brain 137:1282–1303. https://doi.org/10.1093/brain/awt202
Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, Cole FS, Hamvas A, Nogee LM (2014) Genotype–phenotype correlations for infants and children with ABCA3 deficiency. Am J Resp Crit Care Med 189:1538–1543. https://doi.org/10.1164/rccm.201402-0342OC
Kim JH, Park E, Hyun HS, Lee BH, Kim GH, Lee JH, Park YS, Kang HG, Ha IS, Cheong HI (2017) Genotype and phenotype analysis in pediatric patients with cystinuria. J Korean Med Sci 32:310–314. https://doi.org/10.3346/jkms.2017.32.2.310
Mahdavi M, Koulivand L, Khorrami M, Mirsafaie M, Kheirollahi M (2018) In silico analysis of SLC3A1 and SLC7A9 mutations in Iranian patients with cystinuria. Mol Biol Rep. https://doi.org/10.1007/s11033-018-4269-6
Martell HJ, Wong KA, Martin JF, Kassam Z, Thomas K, Wass MN (2017) Associating mutations causing cystinuria with disease severity with the aim of providing precision medicine. BMC Genom 18(Suppl 5):550. https://doi.org/10.1186/s12864-017-3913-1
Wong KA, Wass M, Thomas K (2016) The role of protein modelling in predicting the disease severity of cystinuria. Eur Urol 69:543–544. https://doi.org/10.1016/j.eururo.2015.10.039
Tostivint I, Royer N, Nicolas M, Bourillon A, Czerkiewicz I, Becker PH, Muller F, Benoist JF (2017) Spectrum of mutations in cystinuria patients presenting with prenatal hyperechoic colon. Clin Genet 92:632–638. https://doi.org/10.1111/cge.13079
Kato T (1977) Renal handling of dibasic amino acids and cystine in cystinuria. Clin Sci Mol Med 53:9–15
Stephens AD, Perrett D (1976) Cystinuria: a new genetic variant. Clin Sci Mol Med 51:27–32
Brodehl J, Gellissen K, Kowalewski S (1967) Isolated cystinuria (without lysin-, ornithin and argininuria) in a family with hypocalcemic tetany. Monatsschrift fur Kinderheilkunde 115:317–320
Chillaron J, Roca R, Valencia A, Zorzano A, Palacin M (2001) Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Physiol Renal Physiol 281:F995–F1018. https://doi.org/10.1152/ajprenal.2001.281.6.F995
Fernandez E, Carrascal M, Rousaud F, Abian J, Zorzano A, Palacin M, Chillaron J (2002) rBAT-b(0,+)AT heterodimer is the main apical reabsorption system for cystine in the kidney. Am J Physiol Renal Physiol 283:F540–F548. https://doi.org/10.1152/ajprenal.00071.2002
Feliubadalo L, Arbones ML, Manas S, Chillaron J, Visa J, Rodes M, Rousaud F, Zorzano A, Palacin M, Nunes V (2003) Slc7a9-deficient mice develop cystinuria non-I and cystine urolithiasis. Hum Mol Genet 12:2097–2108. https://doi.org/10.1093/hmg/ddg228
Di Giacopo A, Rubio-Aliaga I, Cantone A, Artunc F, Rexhepaj R, Frey-Wagner I, Font-Llitjos M, Gehring N, Stange G, Jaenecke I, Mohebbi N, Closs EI, Palacin M, Nunes V, Daniel H, Lang F, Capasso G, Wagner CA (2013) Differential cystine and dibasic amino acid handling after loss of function of the amino acid transporter b0,+AT (Slc7a9) in mice. Am J Physiol Renal Physiol 305:F1645–F1655. https://doi.org/10.1152/ajprenal.00221.2013
Nagamori S, Wiriyasermkul P, Guarch ME, Okuyama H, Nakagomi S, Tadagaki K, Nishinaka Y, Bodoy S, Takafuji K, Okuda S, Kurokawa J, Ohgaki R, Nunes V, Palacin M, Kanai Y (2016) Novel cystine transporter in renal proximal tubule identified as a missing partner of cystinuria-related plasma membrane protein rBAT/SLC3A1. Proc Natl Acad Sci 113:775–780. https://doi.org/10.1073/pnas.1519959113
Ercolani M, Sahota A, Schuler C, Yang M, Evan AP, Reimer D, Barone JG, Tischfield JA, Levin RM (2010) Bladder outlet obstruction in male cystinuria mice. Int Urol Nephrol 42:57–63. https://doi.org/10.1007/s11255-009-9597-y
Livrozet M, Vandermeersch S, Mesnard L, Thioulouse E, Jaubert J, Boffa JJ, Haymann JP, Baud L, Bazin D, Daudon M, Letavernier E (2014) An animal model of type A cystinuria due to spontaneous mutation in 129S2/SvPasCrl mice. PloS One 9:e102700. https://doi.org/10.1371/journal.pone.0102700
Peters T, Thaete C, Wolf S, Popp A, Sedlmeier R, Grosse J, Nehls MC, Russ A, Schlueter V (2003) A mouse model for cystinuria type I. Hum Mol Genet 12:2109–2120. https://doi.org/10.1093/hmg/ddg189
Espino M, Font-Llitjos M, Vilches C, Salido E, Prat E, Lopez de Heredia M, Palacin M, Nunes V (2015) Digenic inheritance in cystinuria mouse model. PloS One 10:e0137277. https://doi.org/10.1371/journal.pone.0137277
Fattah H, Hambaroush Y, Goldfarb DS (2014) Cystine nephrolithiasis. Transl Androl Urol 3:228–233. https://doi.org/10.3978/j.issn.2223-4683.2014.07.04
Zisman AL, Evan AP, Coe FL, Worcester EM (2015) Do kidney stone formers have a kidney disease? Kidney Int 88:1240–1249. https://doi.org/10.1038/ki.2015.254
Slavkovic A, Radovanovic M, Siric Z, Vlajkovic M, Stefanovic V (2002) Extracorporeal shock wave lithotripsy for cystine urolithiasis in children: outcome and complications. Int Urol Nephrol 34:457–461
Gurdal M, Ayyildiz A, Huri E, Kanberoglu H, Karaman MI (2003) A huge bladder cystine stone. Int Urol Nephrol 35:497–499
O’Regan S, Robitaille P, Mongeau JG, Homsy Y (1980) Cystine calcium bladder calculus in a 2-year-old child. J Urol 123:770
Mizukami K, Raj K, Osborne C, Giger U (2016) Cystinuria associated with different SLC7A9 gene variants in the cat. PloS One 11:e0159247. https://doi.org/10.1371/journal.pone.0159247
Salido EC, Li XM, Lu Y, Wang X, Santana A, Roy-Chowdhury N, Torres A, Shapiro LJ, Roy-Chowdhury J (2006) Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc Natl Acad Sci 103:18249–18254. https://doi.org/10.1073/pnas.0607218103
Font-Llitjos M, Feliubadalo L, Espino M, Cleries R, Manas S, Frey IM, Puertas S, Colell G, Palomo S, Aranda J, Visa J, Palacin M, Nunes V (2007) Slc7a9 knockout mouse is a good cystinuria model for antilithiasic pharmacological studies. Am J Physiol Renal Physiol 293:F732–F740. https://doi.org/10.1152/ajprenal.00121.2007
Hu L, Yang Y, Aloysius H, Albanyan H, Yang M, Liang JJ, Yu A, Shtukenberg A, Poloni LN, Kholodovych V, Tischfield JA, Goldfarb DS, Ward MD, Sahota A (2016) l-Cystine diamides as l-cystine crystallization inhibitors for cystinuria. J Med Chem 59:7293–7298. https://doi.org/10.1021/acs.jmedchem.6b00647
Lee MH, Sahota A, Ward MD, Goldfarb DS (2015) Cystine growth inhibition through molecular mimicry: a new paradigm for the prevention of crystal diseases. Curr Rheumatol Rep 17:33. https://doi.org/10.1007/s11926-015-0510-7
Poloni LN, Zhu Z, Garcia-Vazquez N, Yu AC, Connors DM, Hu L, Sahota A, Ward MD, Shtukenberg AG (2017) Role of molecular recognition in l-cystine crystal growth inhibition. Cryst Growth Design 17:2767–2781. https://doi.org/10.1021/acs.cgd.7b00236
Sahota A, Parihar JS, Capaccione KM, Yang M, Noll K, Gordon D, Reimer D, Yang I, Buckley BT, Polunas M, Reuhl KR, Lewis MR, Ward MD, Goldfarb DS, Tischfield JA (2014) Novel cystine ester mimics for the treatment of cystinuria-induced urolithiasis in a knockout mouse model. Urology 84:1249.e1249–1249.e1215. https://doi.org/10.1016/j.urology.2014.07.043
Shim M, Park HK (2014) Multimodal treatments of cystine stones: an observational, retrospective single-center analysis of 14 cases. Korean J Urol 55:515–519. https://doi.org/10.4111/kju.2014.55.8.515
Mandal T, Ward MD (2013) Determination of specific binding interactions at L-cystine crystal surfaces with chemical force microscopy. J Am Chem Soc 135:5525–5528. https://doi.org/10.1021/ja401309d
Rimer JD, An Z, Zhu Z, Lee MH, Goldfarb DS, Wesson JA, Ward MD (2010) Crystal growth inhibitors for the prevention of l-cystine kidney stones through molecular design. Science 330:337–341. https://doi.org/10.1126/science.1191968
Shtukenberg AG, Zhu Z, An Z, Bhandari M, Song P, Kahr B, Ward MD (2013) Illusory spirals and loops in crystal growth. Proc Natl Acad Sci 110:17195–17198. https://doi.org/10.1073/pnas.1311637110
Yang Y, Albanyan H, Lee S, Aloysius H, Liang JJ, Kholodovych V, Sahota A, Hu L (2018) Design, synthesis, and evaluation of l-cystine diamides as l-cystine crystallization inhibitors for cystinuria. Bioorg Med Chem Lett. https://doi.org/10.1016/j.bmcl.2018.03.024
Claes DJ, Jackson E (2012) Cystinuria: mechanisms and management. Pediatr Nephrol 27:2031–2038. https://doi.org/10.1007/s00467-011-2092-6
Coe FL, Evan A, Worcester E (2005) Kidney stone disease. J Clin Invest 115:2598–2608
Masotti A, Laurenzi C, Boenzi S, Pastore A, Taranta A, Bellomo F, Muraca M, Dionisi-Vici C, Bertucci P, Dello Strologo L, Emma F (2014) Gender-related effects on urine l-cystine metastability. Amino Acids 46:415–427. https://doi.org/10.1007/s00726-013-1631-9
Aggarwal KP, Narula S, Kakkar M, Tandon C (2013) Nephrolithiasis: molecular mechanism of renal stone formation and the critical role played by modulators. Biomed Res Int 292953:21. https://doi.org/10.1155/2013/292953
Baumann JM, Affolter B (2014) From crystalluria to kidney stones, some physicochemical aspects of calcium nephrolithiasis. World J Nephrol 3:256–267
Khan SR, Pearle MS, Robertson WG, Gambaro G, Canales BK, Doizi S, Traxer O, Tiselius H-G (2017) Kidney stones. Nat Rev Dis Primers 2:16008. https://doi.org/10.1038/nrdp.2016.8
Rimer JD, Kolbach-Mandel AM, Ward MD, Wesson JA (2017) The role of macromolecules in the formation of kidney stones. Urolithiasis 45:57–74. https://doi.org/10.1007/s00240-016-0948-8
Ratkalkar VN, Kleinman JG (2011) Mechanisms of Stone Formation. Clin Rev Bone Miner Metabol 9:187–197. https://doi.org/10.1007/s12018-011-9104-8
Wesson JA, Ganne V, Beshensky AM, Kleinman JG (2005) Regulation by macromolecules of calcium oxalate crystal aggregation in stone formers. Urol Res 33:206–212. https://doi.org/10.1007/s00240-004-0455-1
Funding
This work was supported in part by the Human Genetics Institute of New Jersey and by NIH Grant R01DK112782.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Goldfarb has received consulting fees from Retrophin. The other authors declare that they have no relevant financial interests. The work described in this manuscript on cystine diamides has been licensed by Rutgers University to PharmaKrysto. Rutgers University is the assignee of Patent Number US 9,248,453 B2 entitled “Cystine diamide analogs for the prevention of cystine stone formation in cystinuria” and New York University is the assignee of Patent Number US 8,450,089 B2 entitled “Compounds as L-cystine crystallization inhibitors and uses thereof”.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the Institutional Animal Care and Use Committee of Rutgers University.
Rights and permissions
About this article

Cite this article
Sahota, A., Tischfield, J.A., Goldfarb, D.S. et al. Cystinuria: genetic aspects, mouse models, and a new approach to therapy. Urolithiasis 47, 57–66 (2019). https://doi.org/10.1007/s00240-018-1101-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00240-018-1101-7