
Abstract
Cystinuria is a genetic cause of kidney stones with a prevalence of 1 in 7000 births. So far, two genes have been described responsible for this disorder (SLC3A1 and SLC7A9). We report a patient with an SLC7A9 gene mutation located in 19q13.1 on one allele and with a 19q12q13 region deletion on the other allele. The characteristic clinical features of the 19q13.1 microdeletion syndrome include facial dysmorphism, signs of ectodermal dysplasia, growth retardation, neurologic features and genitourinary anomalies. Cystinuria has not yet been described as part of this syndrome, although one of its responsible genes (SLC7A9) is in the same genomic location. The index case is a 6-year-old male presented with distinctive facial features, cutis aplasia of the scalp, rudimentary teeth, microcephaly, intrauterine and postnatal growth retardation, psychomotor developmental delay, speech delay, epilepsy, inguinal hernias and cystinuria. An array-CGH analysis was performed, finding a large deletion of the 19q12q13.11 cytobands, which affects 19 genes. Two of them are involved in the 19q13.11 deletion syndrome and another affected gene is SLC7A9, responsible for type B cystinuria. Sanger sequencing was performed as well, detecting a heterozygous mutation of the SLC7A9 gene, located in 19q13.1. As far as we know, this is the first described case of cystinuria in a patient with SLC7A9 gene mutation located in 19q13.1 on one allele and with 19q12q13 region deletion on the other allele. Although this patient can be classified as a type B heterozygote and, therefore, his renal prognosis is not severe, the occasional nephrolithiasis found in such patients justifies a close follow-up with regular testing of urinary cystine excretion. We suggest that the recessive behavior of this case, explains the clinical features regarding cystinuria. We propose that in the face of patients affected of a phenotype matchable with 19q13.11 syndrome and cystinuria, a mutational or sequencing study of the SLC7A9 gene should be performed to allow an early onset of diagnosis and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Case report
The index case is a 6-year-old male affected of a rare and complex genetic syndrome, the 19q13.11 deletion syndrome, which includes facial dysmorphism, signs of ectodermal dysplasia, growth retardation, neurologic features and genitourinary anomalies [1–4].
Family history of our patient is unremarkable. He has non-consanguineous healthy parents and two older half-sisters (on his mother’s side) who are also healthy. He was born after an IVF process and presented harmonic intrauterine growth retardation. He remained hospitalized over a month in NICU because of his low weight (1120 g at 34 weeks of gestation, −3.69 SD). When he was 2 months old, he needed repair surgery for bilateral inguinal hernias. He presented a catch up growth, achieving a normal height percentile when he was 1 year old and normal weight when he was four. His head circumference has also improved over the years, but is still to be considered as microcephaly (−2 SD).
His distinctive facial features are low-set ears, broad nasal bridge, arched eyebrows, short palpebral fissures and smooth philtrum (Fig. 1). His signs of ectodermal dysplasia are cutis aplasia of the scalp and malpositioned rudimentary teeth.

Distinctive facial features of our patient. Note low-set ears, broad nasal bridge, arched eyebrows, short palpebral fissures and smooth philtrum. Also note malpositioned rudimentary teeth
His neurological symptoms were first noticed when he was 7 months old as a lack of eye fixation caught the pediatrician’s attention. Only a month later, he had his first epileptic seizure and an electroencephalography (EEG) was made. The diagnosis of the neuropediatric team was “secondarily generalized partial seizures and mild psychomotor developmental delay”.
His parents started a pilgrimage through hospitals in which several studies were made but no underlying cause could be found. He had a normal cranial MRI; several normal EEGs; a normal ophthalmology study which included visually evoked potentials; a normal karyotype study (46, XY); and normal muscle and cutaneous biopsies. A meticulous screening for heredodegenerative disorders was also made, studying neurotransmitters and 5-methyltetrahydrofolate in spinal fluid, SAICAR test, CDT percentage and biotinidase. Amino acids and organic acids blood and urine studies were also performed, with the only remarkable finding of elevated cystine and lysine urine levels.
His epileptic seizures have never been totally controlled, despite the use and combination of different antiepileptic drugs such as oxcarbazepine, valproic acid, levetiracetam and topiramate. He has been hospitalized 16 times because of the seizures, staying at ICU once due to an epileptic status. At the present time, he is enduring 2–4 seizures/month. He is attending a special education school where he improves his intellectual disabilities and lack of communication.
Parallely, when our patient was 13 months old, he suffered a febrile urinary tract infection caused by Klebsiella. He subsequently was studied, finding a II–III grade vesicoureteral reflux with no evidence of obstructive uropathy. A renal gammagraphy was performed 6 months later with normal results. He received prophylactic antibiotic treatment with trimethoprim for a year. He has never presented a urinary tract infection ever since. A renal ultrasonography has been performed annually, with normal results. Renal function and blood pressure have also been regularly assessed.
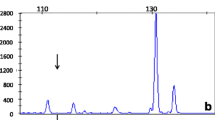
Cystine and lysine urine levels have been repeatedly mildly–moderately elevated. Suspecting this finding to be part of a larger genetic syndrome, an array-CGH analysis was made when our patient was 3 years old. 60,000 oligonucleotides were used, achieving an analysis resolution of 200 kb and finding a deletion of the 19q12q13.11 cytobands. This large deletion (5.81 Mb), considered to be causal, affects 19 genes listed in the OMIM database. Two of them are LSM14A and UBA2, which are involved in the 19q13.11 deletion syndrome [5]. The genomic formula following the ISCN 2009 nomenclature is “arr 19q12q13.11(29738392-35549776)x1”. Another affected gene is SLC7A9, responsible for the cationic amino acid transporter and accordingly causing type B cystinuria [6].
Discussion
Cystinuria is a genetic cause of kidney stones due to decreased proximal tubular reabsorption of filtered cystine performed by the cystine transporter [7, 8]. Two genes have been described to be responsible for this disorder, SLC3A1 and SLC7A9 [6]. SLC3A1 has already been associated with a neurological syndrome: the hypotonia–cystinuria syndrome [9]. We report a defect in SLC7A9 located in 19q13.1 on one allele and with deletion of the 19q12q13 region in the other allele, as part of a complex syndrome: the 19q13.11 microdeletion syndrome. Our patient presents all characteristic clinical features of this syndrome, which include facial dysmorphism, signs of ectodermal dysplasia, growth retardation, neurologic features and genitourinary anomalies [1–3]. Cystinuria had not yet been described as part of this syndrome, despite SLC7A9 being in the same genomic location [6].
This patient being a heterozygote, we expect a good renal prognosis. Such patients usually do not form stones because the cystine concentration remains within the range of solubility [10]. Nevertheless, occasional nephrolithiasis can be found in these patients, justifying a close follow-up with regular testing of urinary cystine excretion.
We suggest that the recessive behavior of this case explains the clinical features regarding cystinuria. The phenotypic characteristics of the patient correlate with the 19q13.11 syndrome.
We propose that in the face of patients affected of a phenotype matchable with 19q13.11 syndrome and cystinuria, a mutational or sequencing study of the SLC7A9 gene should be performed, to allow an early onset of diagnosis and treatment.
References
Chowdhury S, Bandholz AM, et al. Phenotypic and molecular characterization of 19q12q13.1 deletions: a report of five patients. Am J Med Genet Part A. 2014;164A:62–9.
Gana S, Veggiotti P, et al. 19q13.11 cryptic deletion: description of two new cases and indication for a role of WTIP haploinsufficiency in hypospadias. Eur J Hum Genet. 2012;20(8):852–6.
Malan V, Raoul O, et al. 19q13.11 deletion syndrome: a novel clinically recognisable genetic condition identified by array comparative genomic hybridisation. J Med Genet. 2009;46(9):635–40.
Schuurs-Hoeijmakers JH, Vermeer S, et al. Refining the critical region of the novel 19q13.11 microdeletion syndrome to 750 kb. J Med Genet. 2009;46(6):421–3.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore. MIM Number: 613026:2009. http://omim.org/.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore. MIM Number: 220100:2012. http://omim.org/.
Chillarón J, Font-Llitjós M, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6:424.
Claes DJ, Jackson E. Cystinuria: mechanisms and management. Pediatr Nephrol. 2012;27(11):2031–8.
Eggermann T, Spengler S, Venghaus A, et al. 2p21 Deletions in hypotonia-cystinuria syndrome. Eur J Med Genet. 2012;55(10):561–3.
Elkoushy MA, Andonian S. Characterization of patients with heterozygous cystinuria. Urology. 2012;80(4):795–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
About this article

Cite this article
de Rojas, T., Aparicio, C., de Lucas, C. et al. Cystinuria in a patient with 19q12q13.1 deletion. CEN Case Rep 5, 67–69 (2016). https://doi.org/10.1007/s13730-015-0193-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-015-0193-y