
Abstract
In birds the major histocompatibility complex (MHC) organization differs both among and within orders; chickens Gallus gallus of the order Galliformes have a simple arrangement, while many songbirds of the order Passeriformes have a more complex arrangement with larger numbers of MHC class I and II genes. Chicken MHC genes are found at two independent loci, classical MHC-B and non-classical MHC-Y, whereas non-classical MHC genes are yet to be verified in passerines. Here we characterize MHC class I transcripts (α1 to α3 domain) and perform amplicon sequencing using a next-generation sequencing technique on exon 3 from house sparrow Passer domesticus (a passerine) families. Then we use phylogenetic, selection, and segregation analyses to gain a better understanding of the MHC class I organization. Trees based on the α1 and α2 domain revealed a distinct cluster with short terminal branches for transcripts with a 6-bp deletion. Interestingly, this cluster was not seen in the tree based on the α3 domain. 21 exon 3 sequences were verified in a single individual and the average numbers within an individual were nine and five for sequences with and without a 6-bp deletion, respectively. All individuals had exon 3 sequences with and without a 6-bp deletion. The sequences with a 6-bp deletion have many characteristics in common with non-classical MHC, e.g., highly conserved amino acid positions were substituted compared with the other alleles, low nucleotide diversity and just a single site was subject to positive selection. However, these alleles also have characteristics that suggest they could be classical, e.g., complete linkage and absence of a distinct cluster in a tree based on the α3 domain. Thus, we cannot determine for certain whether or not the alleles with a 6-bp deletion are non-classical based on our present data. Further analyses on segregation patterns of these alleles in combination with dating the 6-bp deletion through MHC characterization across the genus Passer may solve this matter in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) is a highly polymorphic gene complex that encodes MHC class I (MHC-I) and II (MHC-II) molecules which are of central importance in the acquired immune system of all vertebrates (Murphy et al. 2008). Classical MHC-I molecules are expressed on all nucleated cells and inhabit receptor structures that bind short peptides (antigens) derived from intracellular pathogens (like viruses) as well as an individual’s own body. After an antigen has been bound, the MHC–antigen complex is transported to the cell surface where it is recognized by CD8+ T cells. When the presented antigen is from a pathogen CD8+ T cell become activated and the infected host cell is killed (Murphy et al. 2008). Non-classical MHC-I molecules (a.k.a. class-Ib) have a similar structure to classical class I molecules but they are less polymorphic and are not expressed to the same extent (Shawar et al. 1994). Moreover, MHC class Ib molecules can have different functions to classical MHC and even bind non-peptide antigens (Rodgers and Cook 2005).
During the last decades evolutionary biologists have been fascinated by how selection maintains polymorphism in classical MHC genes (Hughes and Nei 1988; Potts and Wakeland 1990; Hedrick 2002; Borghans et al. 2004). Evidence that these genes are subject to balancing selection is convincing, e.g., high allelic diversity and heterozygosity as well as an even distribution of alleles within populations (Doherty and Zinkernagel 1975; Hughes and Yeager 1998; Hedrick 1994). Balancing selection is a special case of positive selection whereby a large number of alleles are maintained over long time periods. This can potentially result in identical alleles across species that pre-dated the split of closely related species, i.e., trans-species evolution (Yeager and Hughes 1996; Bos and Waldman 2006). Another example of evidence for positive selection is when the rate of non-synonymous substitutions (d N) is higher than the rate of synonymous substitution (d S) (Hughes and Nei 1988). Mutations that alter amino acids (d N) are favored when they occur in the peptide binding region (PBR) of MHC genes and a d N/d S ratio of greater than one in the PBR is characteristic of classical MHC genes.
In birds MHC organization differs both among and within bird orders. For example, in chickens Gallus gallus of the order Galliformes the entire MHC region is less than 100 kb, it has a simple arrangement with few pseudo-genes, short introns and only one MHC-I and IIB gene is expressed to a high level—referred to as “minimal essential” (Kaufman et al. 1995, 1999). The two MHC-I and MHC-IIB genes in chickens are found at two independent loci, at MHC-B where the alleles have classical MHC characteristics and at MHC-Y where the alleles lack most classical MHC characteristics and are transcribed to a lower extent (Miller et al. 1996; Delany et al. 2009). Two MHC-IIB loci have been reported in additional Galliforme birds, e.g., black grouse Tetrao tetrix and ring-necked pheasants Phasianus colchicus (Wittzell et al. 1999; Strand et al. 2007). However, the MHC organization in another Galliforme, the Japanese quail Coturnix japonica, is much more flexible and diverse. Japanese quail have at least seven class I and ten class IIB loci, although not all loci are transcribed (Shiina et al. 2004; Hosomichi et al. 2006). The green-rumped parrotlet Forpus passerines of the order Psittaciformes has a single highly polymorphic MHC-IIB locus and birds of the order Falconiformes are thought to have between one and two MHC-I locus/loci (Hughes et al. 2008; Alcaide et al. 2009). In passerines (songbirds) of the order Passeriformes, the number of MHC-I and MHC-IIB loci are generally larger than in Galliformes (reviewed in Westerdahl 2007; Promerova et al. 2009; Bollmer et al. 2010; Zagalska-Neubauer et al. 2010; Schut et al. 2011; Sepil et al. 2012) and the passerine MHC organization is more complex with many gene copies and pseudo-genes (Hess et al. 2000; Balakrishnan et al. 2010; Ekblom et al. 2010, 2011).
During the past 10 years MHC-I genes, partial to more complete, have been DNA sequenced in a handful of passerines; great reed warblers Acrocephalus arundinaceus (Westerdahl et al. 1999), scarlet rose finches Carpodacus erythrinus (Promerova et al. 2009), house sparrows Passer domesticus, (Bonneaud et al. 2004), zebra finches, Taeniopygia guttata (Balakrishnan et al. 2010; Ekblom et al. 2010), and blue tits Cyanistes caeruleus (Schut et al. 2011). These five passerines have between at least two and eight MHC-I loci. However, several studies, using restriction fragment length polymorphisms (RFLPs) and Southern Blot techniques, suggest that additional passerines may actually have many more MHC-I loci (Wittzell et al. 1998; Westerdahl 2007). A recent study by Sepil et al. (2012) confirms this suggestion using amplicon 454-sequencing in great tits Parus major. Still, there is very limited knowledge regarding the expression level, function, and structure of MHC genes in passerines. As mentioned previously, birds of the order Galliformes have MHC genes that segregate independently, MHC-B and MHC-Y. Preliminary results (based on RFLP and Southern Blot) from a passerine, the Savannah sparrows Passerculus sandwichensis, suggest that these birds have two independently segregating MHC-IIB loci (Freeman-Gallant et al. 2002), but see Westerdahl et al. (2004). This finding suggests that passerines may also have two different MHC genes—potentially with different functions. Earlier studies conducted on another passerine, the house sparrow, indicate that they have a subset of MHC-I genes, with a 6-bp deletion, that potentially show non-classical characteristics. Firstly, in phylogenetic analyses the MHC-I exon 3 sequences with a 6-bp deletion form a monophyletic cluster with short terminal branches which is in contrast with classical MHC-I exon 3 sequences that often have poor resolution and long terminal branches (Bonneaud et al. 2004). Secondly, exon 3 sequences with a 6-bp deletion have low nucleotide diversity and no positively selected sites (Borg et al. 2011).
Researchers often amplify MHC-I exon 3 and MHC-IIB exon 2 when aiming to study selection acting on the PBR in non-model species. However, in a time where next generation sequencing makes it possible to screen thousands of individuals in a day (e.g., using amplicon 454-sequencing) we think that it is critical to initially partly characterize the MHC genes in non-model study species before deciding which loci and what part of the gene to focus on. This may be particularly important in studies that correlate aspects of MHC genes with traits that influence fitness, such as disease resistance. Knowledge from full-length cDNA and large scale genome projects are essential to better understand how MHC has evolved in passerines, and for us to know which MHC loci are most relevant to screen in ecological study systems. In the present study we will investigate MHC-I in house sparrows in detail to gain a better understanding of putatively classical and non-classical genes in passerines. We use cDNAs and infer the whole PBR which consists of two domains, α1 and α2 (corresponding to exon 2 and 3). We also analyze the α3 domain, corresponding to exon 4 which, e.g., encodes the CD8+ T cell binding site. Working with the α1 to α3 domains, approximately 800 bp, will provide a more rigid analysis for the potential existence of two different MHC-I genes in house sparrows. We then performed amplicon 454-sequencing on exon 3 from genomic DNA, separately for genes with and without the 6-bp deletion, and analyze recombination, selection and linkage.
Materials and Methods
Study Population
The study population was house sparrows inhabiting Lundy Island (51°10′N, 4°40′W, UK) which is a small island (445 hectares) in the Bristol Channel. They are an isolated population with almost no immigration or emigration and the life history of each bird is well documented (Nakagawa and Burke 2008, Nakagawa et al. 2008; Cleasby et al. 2010; Schroeder et al. 2012). The population was established in the seventies but during the winter 1996–1997 the population almost went extinct due to rat poisoning. After this severe bottleneck the population did not recover properly and in 2000, 33 males and 17 females were introduced from the mainland (Ockendon et al. 2009). The population began to increase in size and since 2003 it has fluctuated between 40 and 80 breeding pairs per year (Cleasby et al. 2010). After the introduction the population has been subject to systematic monitoring, with almost all breeding birds and fledglings being marked individually with colour ring combinations in addition to a metal ring supplied by the British Trust for Ornithology (BTO). Almost all birds breed in nest boxes which enables a complete record of all reproductive attempts (Nakagawa and Burke 2008; Nakagawa et al. 2008; Cleasby et al. 2010; Schroeder et al. 2012). The present study is based on genomic DNA samples from 45 individuals in seven families (eight males, seven females, and 30 nestlings) and RNA samples from two additional individuals from the Lundy population.
Isolation of DNA and RNA
Blood samples of 20–40 μl were taken from the brachial vein of adult individuals and 12-day-old chicks captured with mist net, walking trap and in the nest boxes. The blood for DNA extraction was stored in either 100 % ethanol or 500 μl of SET-buffer (150 mM NaCl, 50 mM TRIS, 1 mM EDTA, pH 8.0). Genomic DNA was extracted using salt extraction or standard phenol/chloroform–isoamylalcohol extraction (Sambrook et al. 1989). For isolation of RNA, blood was collected in 100 μl K2EDTA (0.2 M) and then 500 μl TRIzol-LS (Invitrogen, Paisley, UK) was added. Isolation of total RNA was performed using a trizol/chloroform extraction protocol: Blood samples were homogenized in a total volume of 1 ml TRIzol-LS, incubated for 5 min in room temperature and then 200 μl of chloroform was added and then incubated for 3 min. After vortexing for 15 s the samples were incubated for 3 min and then centrifuged at 11,000 rpm for 15 min at 4 °C. The supernatant was transferred to new tubes and 500 μl of isopropanol was added. The samples were inverted 6–7 times and left to rest at room temperature for 30 min before being centrifuged at 11,000 rpm for 10 min at 4 °C. The supernatant was removed, the pellet was washed in ice cold 70 % ethanol and then the sample was centrifuged at 8,500 rpm for 5 min at 4 °C. The ethanol was removed and the pellet was left to air dry. The pellet was dissolved in 0.5 μl RNasin Plus RNase inhibitor (Promega, USA) and 40 μl of ddH2O before being stored in the freezer (−80 °C).
Amplification of MHC-I Exon 3
Genomic exon 3 sequences were amplified from two individuals using degenerate primers; forward primers Pado 10 (5′-TYTCCACACACMTGGTTGCGAG-3′) and A21B (Bonneaud et al. 2004) and reverse primer A23H3 (Balakrishnan et al. 2010), in a standard polymerase chain reaction (PCR) following the protocol for the AmpliTaq polymerase kit (Applied biosystems, New Jersey, US) using 25 ng of genomic DNA. The reaction was run in a thermal cycler GeneAmp PCR system 9700 beginning with an initial denaturation at 94 °C for 2 min, followed by 35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 45 s and finally an extension period of 10 min. The PCR products were checked on a 2 % agarose gel using electrophoresis in 0.5× TBE buffer (890 mM Tris, 890 mM boric acid, and 20 mM EDTA pH 8.0). PCR products (approximately 230 bp, primers not included) from the two individuals were then ligated into a vector and transformed into bacteria using the 2.1 TOPO-TA cloning kit (Invitrogen, US) according to the manufacturer’s protocol. 24 positive (white) colonies were picked from each individual and put in 100 μl of ddH2O. The colonies were heated at 98 °C for 3 min and then put on ice. A new PCR was performed with M13 forward and M13 reverse primers (T m 50 °C) from the cloning kit (Invitrogen, US). Colonies containing inserts of the correct length were identified on an agarose gel, 6 μl of the PCR products (8–12 clones per individual) was purified using 1 μl ExoSap-IT (USB corporation, US) by incubating at 37 °C for 15 min, 80 °C for 15 min, and at 10 °C for at least 10 min and finally 2 μl of this purified product was used as template in a BigDye terminator sequencing reaction using BigDye terminator kit v.3.1. Sequences were obtained from an ABI PRISM 3130 genetic analyzer (Applied biosystems, New Jersey, US).
Rapid Amplification of cDNA Ends
Rapid amplification of cDNA ends, RACE, was used to obtain full-length cDNAs.
Primers (1M11, 5′-GCTACGACGGGCGGGATTTCATCTCCTT-3′; 1M12, 5′-GACCTGGAATCCGGGAGATTCGTGGCAG-3′ in the RACE 3′ reaction; 1RACE5, 5′-CTGCCACGAATCTCCCGGATTC-3′; 1RACE5n 5′-TGGGCAGYTGTGCTTCAGGTAATTCGTC-3′ in the 5′ RACE reaction) within exon 3 were designed in a conserved region using the new sequence information obtained from exon 3 (above). These primers were then utilized in RACE reactions using the SMART™ RACE cDNA amplification kit with mRNA from a single individual as a template. Two forward facing primers were used in 3′ RACE (1M11 and 1M12) and two reverse facing primers (1RACE5 and 1RACE5n) were used in the 5′ RACE. Each reaction was performed following the protocol provided for the RACE-kit (Clonetec, Mountain View, CA). The RACE products were cloned and sequenced as described above.
Amplification of MHC-I Exon 2–4
Using the RACE sequence information new primers were designed (padoM2, 5′-GTT CTC CAC TCC CTG CRT YAC CTG-3′ and padoM4, 5′-CA AGC RAA GAT CCC GGG CTC CAG C-3′) that amplify 756–762 bp (primers not included) covering the major part of exon 2–4, the forward primer was situated in the very beginning of exon 2 and the reverse primer in the end of exon 4. RT-PCR on mRNA was run on two Lundy individuals using the RETROscript kit according to the manufactures protocol (RETROscript kit, Ambion, Austin, USA). The retrieved cDNA was stored at −20 °C before being used as templates in PCR. Standard PCRs were run on the cDNA using the primer combinations padoM2 and padoM4, that amplify exon 2–4, and A21B and A23H3 that amplify exon 3 as described above. For the padoM2 and padoM4 PCR there was an initial denaturation at 94 °C, 2 min, followed by 35 cycles at 94 °C for 30 s, 66 °C for 30 s, and 72 °C for 120 s and finally an extension period of 10 min. The PCR products (exon 2–4 and exon 3 PCR products) were cloned and sequenced as above. Sequences found in at least two independent PCRs, in the same individual or in different individuals, are denoted verified and only such sequences are reported in this study.
454-Amplicon Sequencing
New primers were designed based on the novel exon 2–4 sequence information. These primers either amplify exon 3 in MHC-I alleles with a 6-bp deletion so-called ‘short’ [these sequences are 219 bp (primers not included) shortfw3 5′-GTCTMCACACGAGGTTGCGAG-3′ and rv3 5′-TGCGCTCCAGCTCCYTCTGCC-3′], or exon 3 in MHC-I alleles with no deletion/3 bp deletion so-called ‘long’ [these sequences are 222–225 bp (primers not included) longfw2 5′-GTCTCCACACTGTACAGYGGC-3′ and rv3 5′-TGCGCTCCAGCTCCYTCTGCC-3′]. The reverse primer rv3 was the same in both PCRs (Fig. 1). These two primer combinations were tested in standard PCRs before tagged forward and reverse primers were ordered (Babik et al. 2009). Each PCR, 15 μl total volume using QIAGEN Multiplex MasterMix, contained 25–50 ng genomic DNA and 0.2 μΜ of each primer (QIAGEN). The PCR setting for both primer combinations were; 95 °C for 15 min and then 35 cycles of 95 °C for 30 s, 65 °C for 60 s, and 72 °C for 60 s, and a final extension at 72 °C for 10 min. PCR products were run on gels as above and then equal amounts of four samples were pooled and then purified using MinElute PCR purification kit (QIAGEN) according to the manufacturer’s protocol. The concentrations in these pools were measured with a Nanodrop spectrophotometer and adjusted accordingly before being pooled again into four groups in total (corresponding to four regions on the 454-chip). Each region contained 12 individually tagged ‘short’ and 12 ‘long’ MHC-I PCR products. The amount of each sample was adjusted for sequence coverage of 300 reads per individual for each primer combination (‘short’ and ‘long’) in a 454 FLX instrument sequencing run.

Schematic diagram of the positions of the primers used in the present study (MHC-I exon 2, intron 2, exon 3, intron 3 and exon 4). Forward primers are shown above the exons (shortfw3, longfw2, pado10, padoM2, A21B, 1M11, 1M12) and reverse primers below the exons (1RACE5, 1RACE5n, padoM4, rv3, A23H3). All primer sequences are indicated in the text, the primer combination shortfw3-rv3 amplifies exon 3 sequences with the 6-bp deletion and the primer combination longfw2-rv3 amplifies exon 3 sequences without the 6-bp deletion
Filtering of 454 Data
The 454-sequencing data was filtered to remove low-quality sequences, any reads representing artefactual MHC alleles and potentially non-functional alleles. Only sequences with a perfect match to both the forward and reverse primers and with complete 6 bp tags were extracted from the 454 multifasta file using the program jMHC (Stuglik et al. 2011). Unique sequences, in length and nucleotide composition, were then identified and sorted according to the tags using jMHC (Stuglik et al. 2011). Any identical sequences within individuals were detected and merged using the web-applications seqeqseq (http://mbio-serv2.mbioekol.lu.se/apps/seqeqseq.html) and mergeMatrix (http://mbio-serv2.mbioekol.lu.se/apps/mergeMatrix.html). To remove artefactual alleles we used bioinformatic filters (Filter 1, 3, and 4) in the web-application popMatrix (http://mbio-serv2.mbioekol.lu.se/apps/popMatrix.html). (1) Filter 1 (minimum total sequence abundance/individual) was set to 119 reads for the ‘short’ primer combination and to 55 reads for the ‘long’ primer combination (settings according to Galan et al. (2010)), hence the total number of MHC-I exon 3 reads per individual was >173. (2) Technical duplicates of samples (three per primer combination) were used to determine cut-off values for the filtering of verified/false alleles. The Filter 3 (minimum relative abundance of a sequence/individual) cut-off value was set to 2 % for the ‘short’ primer combination and 1.5 % for the ‘long’ primer combination. Novel alleles had to be found in at least two independent PCR reactions to be verified (Filter 4).
Analysis
All sequences were manually aligned based on conserved amino acids and adding as few insertions as possible in the program BioEdit 7.0.9.0 Sequence Alignment Editor (Hall 1999). Neither the three-dimensional structure of a human HLA-A molecule nor the three-dimensional structure of a chicken MHC-I molecule can really provide the exact peptide binding sites of the MHC-I molecule in house sparrows (Bjorkman et al. 1987; Koch et al. 2007). We decided to use the PBR from HLA-A on the house sparrow sequences, since this has been used in earlier passerine MHC studies to calculate d N and d S (Fig. 2; Westerdahl et al. 1999; Bonneaud et al. 2004), and it was recently shown to correlate well with findings from in silico peptide binding predictions of passerine MHC-I molecules (Follin et al. 2013). Phylogenetic reconstruction of house sparrow amino acid sequences was done in RAxML BlackBox, using Maximum Likelihood (ML) and bootstrapping (bt) with 100 replicates (Jones-Taylor_Thornton (JTT) model, standard settings; Stamatakis et al. 2008). Further phylogenetic reconstructions of house sparrow sequences were done based on nucleotides (all nucleotides, synonymous substitutions, and nonsynonymous substitutions) in MEGA 5.05, using full length sequences, exon 2, exon 3, and exon 4 (Tamura et al. 2011). The rate of non-synonymous (d N) and synonymous substitutions (d S) in the PBR and non-PBR was calculated using the Nei–Gojobori method of pairwise comparison with a Jukes-Cantor correction for multiple hits (Nei and Gojobori 1986) in MEGA 5.05, as were segregating sites, nucleotide diversity and estimates of average evolutionary divergence (Tamura et al. 2011). We tested for positively and negatively selected sites in the online web-service of HyPhy (http://www.datamonkey.org/), first taking recombination into account (GARD), then using the REL method to estimate the d N/d S ratio at every codon in the alignment with the REV nucleotide substitution bias model, and the default significance level (Kosakovsky et al. 2005; Delport et al. 2010).

Alignment of house sparrow MHC-I amino acid sequences, covering the α1, α2, and α3 regions (species-specific nomenclature and GenBank accession numbers are used, Pado), in comparison with great reed warblers (Acar cN3, AJ005503; cN15, AJ005505) and chickens (MHC-B, Gaga, HQ141386; MHC-Y NM_001030675), numbered according to full-length chicken MHC-I. Pado-UA*240-242 do not have the 6-bp deletion, Pado-UA*243 has a 3 bp deletion (site 149) and Pado-UA*230-233 have the 6-bp deletion (site 246–247). Identity with sequence Pado-UA*240 is indicated with dots, codons corresponding to the PBR with (P), and the CD8 residues in the α3 region are underlined
Results
MHC-I, the α1 to α3 Domains
We amplified 756–762 bp, covering the major part of MHC-I α1 to α3 domains (exon 2–4) in two house sparrows (252–254 amino acid codons) and it resulted in eight verified MHC-I alleles (Fig. 2; Suppl. Table 1). Sequences Pado-UA*230-233 have a 6-bp deletion at position 146–147 in the α2 region and also 17 unique sites in the α1 to α2 domains compared with the four alleles without the 6-bp deletion Pado-UA*240-243 (α1, 34 M, 61 K, 66C, 68 K, 78D; α2, 81Q, 110-113FSQD, 143H, 153A, 154R, 158L, 171R, 172C, 175R). There are no consistent amino acids that differ between the sequences with and without the 6-bp deletion in the α3 domain. Allele Pado-UA*243 has a 3-bp deletion at position 149 compared with the alleles Pado-UA*240-242. The amino acid sequence of MHC-I genes in house sparrows are similar to that of other birds and possess many conserved amino acid residues; for example, the presence of four cysteine (C) residues at alignment positions 99,162, 200, and 256 which are responsible for the disulfide bridge formation, the histidine (H) and the aspartic acid (D) that form salt bridges at position 29D in α1, and at 91H and 119D in α2, although this aspartic acid (D) is substituted by H in sequence Pado-UA*231. According to Kaufman et al. (1994) there are eight highly conserved amino acid positions known to bind the C- and N-terminal of peptides and the house sparrow sequences cover seven of these eight sites. Six sites were completely conserved in the house sparrow sequences and found at positions 58Y, 83R, 140T, 144W, 157Y, and 169Y, while the seventh position was variable, 143R/143H. The sequences without a 6-bp deletion have an arginine (R) here, while sequences with the 6-bp deletion have an H (Fig. 2). The CD8 binding sites in the α3 domain are known to be highly conserved in mammals and in house sparrows the CD8 regions are located approximately at positions 220–226, 230–232, and 242–244 (Fig. 2; Kaufman et al. 1994; Glaberman et al. 2008). At position 223 within the CD8 region, some house sparrow sequences with the 6-bp deletion (Pado-UA*231 and Pado-UA*232) have the conserved glutamine (Q) substituted by an H.
Trees based on amino acid sequences were interpreted separately for the α1, α2, and α3 domains, by Maximum Likelihood, with chickens (MHC-B and MHC-Y) as outgroups. House sparrow sequences with a 6-bp deletion formed a significantly supported monophyletic cluster (bt = 97, Fig. 3a) in the tree based on the α1 domain and also in tree based on the α2 domain (bt = 97, Fig. 3b). However, in the tree based on the α3 domain all house sparrow sequences formed a single cluster (bt = 94, Fig. 3c) independent of the 6-bp deletion. Two great reed warbler MHC-I sequences were included in each of the three trees as distantly related reference sequences [sparrows and warblers diverged approximately 25MYA (Sibly et al. 2012)]. In the α1 tree the house sparrow sequences were separated from the great reed warbler sequences (bt = 70, Fig. 3a) whereas in the α2 tree house sparrow sequences without the 6-bp deletion were mixed with the great reed warbler sequences (Fig. 3b) indicating trans-species evolution. Finally in the α3 tree the house sparrow sequences were again separated from great reed warbler sequences (bt = 94, Fig. 3c). The pattern that the house sparrow sequences with a 6-bp deletion were found in a separate cluster for exon 2 and 3 but not for exon 4 was also confirmed in nine additional trees based on nucleotides, synonymous substitutions only and non-synonymous substitutions only (Suppl. Fig. 2).

Phylogenetic reconstruction of house sparrow (Pado-UA*230-233 and Pado-UA*240-243) and great reed warbler (Acar cN3, AJ005503; cN15, AJ005505) exon 2, 3, and 4 MHC-I amino acid sequences (α1, α2, and α3 regions), with chicken sequences (MHC-B, Gaga, HQ141386; MHC-Y NM_001030675) as outgroups, using Maximum Likelihood (JTT model, bootstrap (bt) values based on 100 replicates). a Phylogenetic reconstruction of the α1 region where house sparrow sequences with a 6-bp deletion form a significantly supported monophyletic cluster (bt = 97). b Phylogenetic reconstruction of the α2 region where house sparrow sequences with a 6-bp deletion form a significantly supported cluster (bt = 97). c Phylogenetic reconstruction of the α3 region where all house sparrow sequences, with and without the 6-bp deletion, form a single cluster (bt = 94)
454-Sequencing, α2 Domain (Exon 3)
MHC-I exon 3 sequences without a 6-bp deletion (222–225 bp, ‘long’) were successfully amplified in 36 individuals using 454-pyrosequencing (total number of reads = 9,875). After filtering 31 individuals fulfilled our criteria (average number of reads/individual = 220, total number of reads after filtering = 6,805). MHC-I exon 3 sequences with a 6-bp deletion (219 bp, ‘short’) were successfully amplified in 38 individuals using 454-pyrosequencing (total number of reads = 11,009). After filtering 32 individuals fulfilled our filtering criteria (average number of reads/individual = 264, total number of reads after filtering = 7,131). A total of 20 sequences (5.0 ± 1.6 per individual), without the 6-bp deletion, and 38 sequences (9.4 ± 2.7 per individual), with the 6-bp deletions, were verified. The highest total number of exon 3 sequences found in an individual was 21. The maximum number of exon 3 sequences without a 6-bp deletion within an individual was eight and 15 for sequences with a 6-bp deletion.
The cDNA sequences with a 6-bp deletion (Pado-UA*230-233, reported above) had nine unique sites in the α2 domain, that were covered also by the 454 run (Fig. 2, Suppl. Fig. 1), compared with the alleles without the 6-bp deletion (Pado-UA*240-243). When investigating the uniqueness of these conserved sites in the 58 exon 3 sequences from the 454 run the four sites in exon 3, positions 110-113FSQD, remained unique as did the conserved site 143H (Fig. 2, Suppl. Fig. 1), although the four sites downstream in exon 3 where no longer unique (153A, 154R, 158L, 171R, Fig. 2, Suppl. Fig. 1). The number of segregating sites (S) in the PBR and the nucleotide diversity (π) was low in the sequences with a 6-bp deletion compared to estimates for MHC-I in other birds (Table 1; e.g. Westerdahl et al. 1999; Schut et al. 2011; Strandh et al. 2011). The rate of non-synonymous (d N) substitutions in the PBR was twice as high compared to the rate in the nonPBR, but overall d N and d S was again low compared to estimates in other birds. The d N/d S ratio could not be estimated in the PBR alone since there were no synonymous substitutions (d S) in the PBR (Table 1). The 20 house sparrow sequences without the 6-bp deletion have MHC diversity measures that are similar to those found in other birds (SPBR = 24, Snon-PBR = 52, π PBR = 0.210, π non-PBR = 0.067, dNPBR = 0.268, dSPBR = 0.216, dNnon-PBR = 0.054, dSnon-PBR = 0.143; e.g., Westerdahl et al. 1999; Schut et al. 2011; Strandh et al. 2011). The average evolutionary divergence was low for sequences with the 6-bp deletion and it did not change depending on which part of exon 3 was analyzed (Table 2). The within group distance for alleles without the 6-bp deletion was higher than the overall distance, and the between group distance (comparing alleles with and without the 6-bp deletion) was, as might be expected, highest for the PBR.
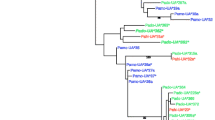
A tree based on the 58 amino acid sequences (α2 domain) from the 454 run was estimated using Maximum Likelihood, with domestic chicken sequences (MHC-B and MHC-Y) as outgroups and sequences from five additional passerines were also added for comparison. All 38 house sparrow sequences with a 6-bp deletion formed a significantly supported monophyletic cluster (bt = 85) with short terminal branches and the four house sparrow sequences with a 3-bp deletion were found spread among house sparrow sequences without the 6-bp deletion. The sequences from five other passerines were found among the house sparrow sequences without the 6-bp deletion, an indication of trans-species evolution, although not significantly so (Fig. 4).

Phylogenetic reconstruction of verified house sparrow exon 3 MHC-I amino acid sequences, from 454 amplicon sequencing (α2 region; 38 sequences with a 6-bp deletion and 20 sequences without this deletion), and exon 3 MHC-I amino acid sequences from additional passerines using Maximum Likelihood (JTT model, bootstrap (bt) values based on 100 replicates). Species-specific nomenclature and GenBank accession numbers are used for all sequences (Pado-UA*200-260 (novel from the present study), Pado-UA*324 JN609623, Pado-UA*326 JN609626, Pado-UA*312 JN609636, Pado-UA*309 JN609635, Pado-UA*322 JN609643, Pado-UA*317 JN609640 (found previously and also in the present study). Also included in the reconstruction are two great reed warbler great reed warbler (Acar cN3, AJ005503; cN15, AJ005505), two Seychelles warbler (Ase_UA9, AJ557882; Ase_UA4, AJ557877), two Scarlet rose finch (Caer_26, JN713048; Caer_93, JN713049), two Berthelot’s Pipit Anthus berthelotii (Anbe_38, JN799612; Anbe_23, JN799619) and one zebra finch (Tagu, XM_002186531) exon 3 sequences with two chicken sequences (MHC-B, Gaga, HQ141386; MHC-Y NM_001030675 as outgroups. House sparrow sequences with a 6-bp deletion form a significantly supported monophyletic cluster (bt = 85), denoted with Pado-UA*xxx_6, while the remaining passerine exon 3 sequences are found among house sparrow sequences without the 6-bp deletion
MHC-I, Recombination and Selection
Recombination events were investigated in the eight house sparrow transcripts and in the 58 exon 3 sequences using HyPhy. There were two significant recombination events at positions 78 and 110 in the long transcripts (Fig. 2), while there were no recombination events in the 58 exon 3 sequences, neither when all 58 sequences where run in a single analysis nor when the sequences with and without the 6-bp deletion were run separately (Suppl. Fig. 1). Taking recombination into account we tested for selection in the eight long house sparrow transcripts and found 42 negatively selected sites (spread out in the entire transcript) but no positively selected sites. We did phylogenetic reconstructions on exon 2 and exon 3 in the eight long transcripts, analyzing the regions upstream, downstream and between the recombination sites separately. The pattern that the house sparrow sequences with a 6-bp deletion were found in a separate cluster was also confirmed in these trees (data not shown). We then performed tests for selection in the exon 3 sequences with the 6-bp deletion (N = 38) and found one positively selected site and 11 negatively selected sites (Suppl. Fig. 1). Subsequently we tested for selection in the sequences without the 6-bp deletion (N = 20) and found four positively selected sites and five negatively selected sites (Suppl. Fig. 1). Finally, when all 58 exon 3 sequences were run in a single analysis there were two positively selected sites and 22 negative (Suppl. Fig. 1). The negatively selected sites were found in certain parts of exon 3 and the regions subject to negative selection are consistent between analyses. However, three out of five positively selected sites disappeared when exon 3 sequences with and without the 6-bp deletion were run in a single analyses compared to when these sequences were run separately.
MHC-I, Segregation of Alleles
Inheritance of house sparrow exon 3 sequences (in this paragraph called alleles), with and without the 6-bp deletion, was studied in three house sparrow families with 2–4 chicks (Suppl. Fig. 3). All individuals carried alleles with and without the 6-bp deletion. We observed segregation of a total of 11 female and 14 male alleles with a 6-bp deletion, and segregation of 7 female and 10 male alleles without the 6-bp deletion (Suppl. Fig. 3). All MHC alleles (with and without the 6-bp deletion) were inherited as two linked haplotypes per parent, hence alleles with and without the 6-bp deletion were completely linked, in our limited data set.
Discussion
The occurrence of both classical and non-classical MHC-I genes is a common phenomenon in vertebrates (Murphy et al. 2008; Delany et al. 2009; Goyos et al. 2011; Reed et al. 2011). However, little is known about these MHC-I genes in passerines, possibly because the genomic MHC region in passerines is complex with many MHC-I and II loci (Westerdahl 2007; Zagalska-Neubauer et al. 2010; Sepil et al. 2012). The “core MHC region” is most likely found on chromosome 16 in the zebra finch (Ekblom et al. 2011), though MHC genes may also be found on additional chromosomes (Balakrishnan et al. 2010). Few studies have reported long transcripts of MHC-I in passerines, information that is valuable for a more comprehensive understanding of the evolution and function of the MHC-I genes (Westerdahl et al. 1999). In the present study we verified eight MHC-I transcripts (α1–α3 region) in house sparrows. Independent phylogenetic reconstruction on the α1 and α2 regions showed that transcripts with a 6-bp deletion (‘short’ alleles) formed a significant monophyletic cluster in both trees (Fig. 3a, b). Additionally, the sequences with a 6-bp deletion had a highly conserved residue in α2 (exon 3) substituted, 143H in place of 143R. This polymorphism in the sequences with a 6-bp deletion was also apparent when a large set of exon 3 sequences from amplicon 454-sequencing was evaluated. Furthermore, the exon 3 sequences with a 6-bp deletion had low nucleotide diversity, a low rate of non-synonymous substitutions and only a single site subject to positive selection. These characteristics are suggestive of non-classical MHC. However, a recent expansion of the allelic lineage with a 6-bp deletion could have generated similar results; a highly supported cluster of sequences with low nucleotide diversity and few positively selected sites. We cannot exclude this latter scenario hence it cannot be concluded whether the house sparrow alleles with a 6-bp deletion are non-classical or not. Some recent data suggests that the alleles with a 6-bp deletion could be rather old. Follin et al. (2013) found MHC-I alleles with a 6-bp deletion in another species from the genus Passer, tree sparrow Passer montanus, indicating that this 6 bp deletion may pre-date the split of these two species, making it around 10 million years old (Sibly et al. 2012). However, it is also possible that the deletion occurred separately in each of these species. Goyos et al. (2011) recently provided convincing evidence of non-classical genes across distantly related Amphibians and a similar approach should be possible to perform in passerines in the near future.
House sparrow transcripts with a 6-bp deletion (Pado-UA*230-233) had several unique sites in the α1 and/or α2 domains, compared with sequences without the 6-bp deletion (Pado-UA*240-243; Fig. 2). The polymorphic site at position 143 may be of particular interest since this site is one of eight highly conserved amino acid positions among vertebrates. In classical MHC-I genes of mammals, chickens, lizards, frogs, and fish lysine (K) is reported at this position (Kaufman et al. 1994). In non-classical MHC-I genes this site is substituted; in the chicken and Galápagos marine iguanas Amblyrhynchus cristatus for R whereas in the turkey Meleagris gallopavo, classical genes have both K and R while non-classical only have R (Kaufman et al. 1994; Glaberman et al. 2008; Reed et al. 2011). In house sparrows R is found in the transcripts without a 6-bp deletion but is substituted for an H in the sequences with the 6-bp deletion (Fig. 2, Suppl. Fig. 1). Surprisingly, K was not found at all in this position in house sparrows and R was found in the genes that we expected to be the more classical MHC-I genes, hence the reversed scenario compared with chicken and marine iguanas. R, H, and K are positively charged hydrophilic amino acids and are found in the same group of amino acids. The functional differences caused by the substitution at position 143 may therefore be of minor importance.
The CD8 domain in α3 is an important binding region for the CD8+T-cells and this region is well conserved within species, whereas it is highly variable between species. However, certain amino acid residues are conserved even between distantly related species, for example, Q at position 223 (Fig. 2) is conserved between mammals, chickens, and lizards (Kaufman et al. 1994, here position 226). Surprisingly this Q is substituted for an H in two of the house sparrow transcripts (Pado-UA*231 and Pado-UA*232, Fig. 2), whereas it is conserved in great reed warblers and chickens MHC-B and MHC-Y. This substitution (from Q to H) has also been found in classical versus non-classical MHC-I in the Galápagos marine iguanas (Glaberman et al. 2008). We only found this substitution in house sparrow MHC-I alleles with the 6-bp deletion (Fig. 2). Q and H are amino acids with different biochemical characteristics and they are negatively and positively charged, respectively. The residues that form the dominant loop for CD8 binding are negatively charged (Salter et al. 1990), thus substitution from a negatively to positively charged amino acid may affect the structure and formation of the CD8 binding site and prevent binding to CD8 (Salter et al. 1989).
In the scarlet rose finch and great reed warbler there are no MHC-I exon 3 sequences that form significantly separate clusters (Westerdahl et al. 1999; Promerova et al. 2009), while such clusters have been reported in blue tits, great tits and house sparrows (Bonneaud et al. 2004; Schut et al. 2011; Sepil et al. 2012; this study). In the current study, the sequences with a 6-bp deletion formed a significant monophyletic cluster with short terminal branches in trees based on both the α1 and α2 regions as well as that based on a larger number of exon 3 amino acid sequences (α2 domain, N = 58; exon 3, Fig. 4, bt = 85). Exon 3 sequences from five additional passerines were included in the later tree and were found among house sparrow sequences without the 6-bp deletions. The clustering in these trees suggests that the alleles with a 6-bp deletion are separated from the rest of the MHC-I alleles. However, a tree based on exon 4 (α3 domain), which encodes the structural part of the MHC-I molecule, showed no separation of house sparrow alleles in relation to the 6-bp deletion. This high sequence similarity in exon 4 is most likely due to purifying selection although gene conversion cannot be excluded as an additional mechanism. Gene conversion has previously been reported between the two MHC-I genes within the classical MHC-B locus for both chicken and turkey (Hosomichi et al. 2008; Reed et al. 2011). Very little is known about the evolutionary history of MHC-I genes in birds, but there is recent knowledge on MHC-IIB and phylogenetic reconstruction on MHC-IIB exon 3, the structural part of the beta chain, revealed that there was a duplication event prior to the major avian radiation resulting in two ancient MHC-IIB lineages (Burri et al. 2010). One of these MHC-IIB lineages was lost in some bird orders and passerine MHC-IIB genes have probably evolved from a single MHC lineage (Burri et al. 2010). If MHC-I have a similar evolutionary history to MHC-IIB then the house sparrow MHC-I genes were duplicated rather recently and exon 4 is then likely to be similar across alleles, with and without the 6-bp deletion, due to purifying selection.
When we investigated genetic diversity and selection in the 38 alleles with the 6-bp deletion these alleles showed characteristic that are different from what is usually seen in passerine MHC genes, i.e., low nucleotide diversity, only a single positively selected site and 11 negatively selected sites (Table 1; Suppl. Fig. 1). The average evolutionary divergence was also low in these sequences, independent of which part of exon 3 was analyzed (Table 2). The genetic diversity and selection in the 20 alleles without the 6-bp deletion showed classic MHC characteristics; high nucleotide diversity, four positively selected site and five negatively selected sites (Table 2; Suppl. Fig. 1). The within group distance for these alleles was higher than the overall distance (including all exon 3 sequences), and the distance was considerably larger in the PBR compared with the non-PBR. Simultaneous selection analyses and d N/d S-ratios on all exon 3 sequences (sequences with and without the 6-bp deletion) proved problematic since the patterns of positive selection were so different in the alleles with a 6-bp deletion. When we did selection analyses on all alleles simultaneously, three out of four previously found positively selected sites in the alleles without the 6-bp deletion disappeared (Suppl. Fig. 1).
There were up to 21 MHC-I exon 3 sequences per individual in total according to our strict filtering criteria of the 454-pyrosequencing data; at least eight loci with exon 3 sequences with a 6-bp deletion (15 exon 3 sequences in a single individual) and at least four loci with without a 6-bp deletion (eight exon 3 sequences in a single individual). We believe that the true number of MHC-I alleles in house sparrows may be even higher since we set the threshold for the minimum number of reads per individual rather low (>173 reads). According to Galan et al. (2010) we would have needed more reads per individual to find every allele three times and with high probability (99.9 %). Previous studies on MHC in house sparrows have underestimated the number of MHC-I loci due to the molecular genetic techniques that were available in the past (e.g., Bonneaud et al. 2004; Borg et al. 2011). If the exon 3 sequences with and without the 6-bp deletion were found in different chromosomal regions, then house sparrows have at least 12 MHC-I loci (8 plus 4 loci). However, the alleles with and without the 6-bp deletion were completely linked when we looked at inheritance in families (Suppl. Fig. 3). This linkage does not contradict that alleles with and without the 6-bp deletion could be found at different chromosomal regions, though it suggests that these regions are linked. This result is preliminary since we only had adequate resolution and high quality 454-pyrosequencing data in a very limited data set, three families (segregation of a total of 42 alleles).
MHC-I exon 2 and 3 encode the peptide binding groove that interacts with antigens (Murphy et al. 2008). Exon 3 has traditionally been the focal exon used for screening MHC-I in non-model organisms. Recent preliminary results on MHC-I in a variety of birds showed that exon 2 was as variable as exon 3 (Strandh et al. 2011). It is therefore important to not only focus on a small part of the MHC molecule initially, when aiming to screen for MHC polymorphism in a non-model organism because we cannot know beforehand which exon is subject to strongest selection and/or is most variable. In the house sparrow we investigated exon 3 since we knew that the 6-bp deletion was found in this exon, although we initially sequenced exon 2, 3, and 4. With knowledge from these exon 2–4 transcripts we were able to design primer pairs that successfully amplified exon 3 sequences with and without the 6-bp deletion (Fig. 1, Suppl. Fig. 1). Today we can easily get a million MHC sequences from amplicon 454-sequencing, however, the output will not be better than the PCR product put into the 454 run. So, when screening MHC variation in a new species, or even a new order, it is appropriate to consider the complex structure of MHC and to sequence at least the main parts of the peptide binding groove before setting up a more narrow screening system.
Many of our findings suggest that alleles with a 6-bp deletion are non-classical MHC-I genes; they form a monophyletic cluster in trees based on both the α1 and α2 region, highly conserved residues are substituted compared with classical MHC genes, they have low nucleotide diversity, only a single site is subject to positive selection and their average evolutionary divergence is low and does not change depending on which part of exon 3 that is analyzed. However, a recent expansion of the allelic lineage with a 6-bp deletion could also have generated these results. Future studies that date the 6-bp deletion could potentially help to determine whether sequences with a 6-bp deletion are non-classical genes or have arisen from a recent expansion.
References
Alcaide M, Edwards SV, Cadahía L, Negro JJ (2009) MHC class I genes of birds of prey: isolation, polymorphism and diversifying selection. Conserv Genet 10:1349–1355
Babik W, Taberlet P, Ejsmond MJ, Radwan J (2009) New generation sequencers as a tool for genotyping of highly polymorphic multilocus MHC system. Mol Ecol Resour 9:713–719. doi:10.1111/j.1755-0998.2009.02622.x
Balakrishnan CN, Ekblom R, Volker M, Westerdahl H, Godinez R, Kotkiewicz H, Burt DW, Graves T, Griffin DK, Warren WC, Edwards SV (2010) Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biol 8:29
Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC (1987) The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329:512–518
Bollmer JL, Dunn PO, Whittingham LA, Wimpee C (2010) Extensive MHC class II B gene duplication in a Passerine, the Common Yellowthroat (Geothlypis trichas). J Hered 101:448–460
Bonneaud C, Sorci G, Morin V, Westerdahl H, Zoorob R, Wittzell H (2004) Diversity of Mhc class I and IIB genes in house sparrows (Passer domesticus). Immunogenetics 55:855–865
Borg AA, Pedersen SA, Jensen H, Westerdahl H (2011) Variation in MHC genotypes in two populations of house sparrow (Passer domesticus) with different population histories. Ecol Evol 1(2):145–159. doi:10.1002/ece3.13
Borghans JAM, Beltman JB, De Boer RJ (2004) MHC polymorphism under host-pathogen coevolution. Immunogenetics 55:732–739
Bos DH, Waldman B (2006) Evolution by recombination and transspecies polymorphism in the MHC class I gene of Xenopus laevis. Mol Biol Evol 23:137–143
Burri R, Salamin N, Studer RA, Roulin A, Fumagalli L (2010) Adaptive divergence of ancient gene duplicates in the avian MHC class II beta. Mol Biol Evol 27:2360–2374
Cleasby IR, Nakagawa S, Gillespie DOS, Burke T (2010) The influence of sex and body size on nestling survival and recruitment in the house sparrow. Biol J Linn Soc 101:680–688
Delany ME, Robinson CM, Goto RM, Miller MM (2009) Architecture and organization of chicken microchromosome 16: order of the NOR, MHC-Y, and MHC-B Subregions. Heredity 100:507–514
Delport W, Poon AF, Frost SDW, Kosakovsky Pond SL (2010) Datamonkey 2010 a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26(19):2455–2457. doi:10.1093/bioinformatics/btq429
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256:50–52
Ekblom R, French L, Slate J, Burke Terry (2010) Evolutionary analysis and expression profiling of zebra finch immune genes. Genome Biol Evol 2:781–790. doi:10.1093/gbe/evq061
Ekblom R, Stapley J, Ball AD, Birkhead T, Burke T, Slate J (2011) Genetic mapping of the major histocompatibility complex in the zebra finch (Taeniopygia guttata). Immunogenetics 63:523–530
Follin E, Karlsson M, Lundegaard C, Nielsen M, Wallin S, Paulsson K, Westerdahl H (2013) In silico peptide-binding predictions of passerine MHC class I reveal similarities across distantly related species, suggesting convergence on the level of protein function. Immunogenetics 65:299–311. doi:10.1007/s00251-012-0676-3
Freeman-Gallant CR, Johnson EM, Saponara F, Stanger M (2002) Variation at the major histocompatibility complex in Savannah sparrows. Mol Ecol 11:1125–1130
Galan M, Guivier E, Caraux G, Charbonnel N, Cosson JF (2010) A 454 multiplex sequencing method for rapid and reliable genotyping of highly variable genes. BMC Genom 2010(11):296
Glaberman S, Du Pasquier L, Caccone A (2008) Characterization of a nonclassical class I MHC gene in a reptile, the Galapagos Marine Iguana (Amblyrhynchus cristatus). PLoS ONE 3(8):e2859. doi:10.1371/journal.pone.0002859
Goyos A, Sowa J, Ohta Y, Robert J (2011) Remarkable conservation of distinct nonclassical MHC class I lineages in divergent amphibian species. J Immunol 186:372–381. doi:10.4049/jimmunol.1001467
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acid Symp Ser 41:95–98
Hedrick PW (1994) Evolutionary genetics of the major histocompatibility complex. Am Nat 143:945–964
Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56:1902–1908
Hess CM, Gasper J, Hoekstra HE, Hill CE, Edwards SV (2000) MHC class II pseudogene and genomic signature of a 32-kb cosmid in the house finch (Carpodacus mexicanus). Genome Res 10:613–623
Hosomichi K, Shiina T, Suzuki S, Tanaka M, Shimizu S, Iwamoto S, Hara H, Yoshida Y, Kulski J, Inoko H, Hanzawa K (2006) The major histocompatibility complex (Mhc) class IIB region has greater genomic structural flexibility and diversity in the quail than the chicken. BMC Genom 7:322. doi:10.1186/1471-2164-7-322
Hosomichi K, Miller MM, Goto RM, Wang Y, Suzuki S, Kulski JK, Nishibori M, Inoko H, Hanzawa K, Shiina T (2008) Contribution of mutation, recombination, and gene conversion to chicken MHC-B haplotype diversity. J Immunol 181:3393–3399
Hughes AL, Nei M (1988) Patterns of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167–170
Hughes AL, Yeager M (1998) Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 32:415–435
Hughes CR, Miles S, Walbroehl JM (2008) Support for the minimal essential MHC hypothesis: a parrot with a single, highly polymorphic MHC class II B gene. Immunogenetics 60:219–231
Kaufman J, Salomonsen J, Flajnik M (1994) Evolutionary conservation of MHC class I and class II molecules—different yet the same. Semin Immunol 6:411–424
Kaufman J, Volk H, Wallny HJ (1995) A “minimal essential Mhc” and an “unrecognized Mhc”: two extremes in selection for polymorphism. Immunol Rev 143:63–88
Kaufman J, Milne S, Gobel TW, Walker BA, Jacob JP, Auffray C, Zoorob R, Beck S (1999) The chicken B locus is a minimal essential major histocompatibility complex. Nature 401:923–925
Koch M, Camp S, Collen T, Avila D, Salomonsen J, Wallny HJ, van Hateren A, Hunt L, Jacob JP, Johnston F, Marston DA, Shaw I, Dunbar PR, Cerundolo V, Jones EY, Kaufman J (2007) Structures of an MHC class I molecule from B21 chickens illustrate promiscuous peptide binding. Immunity 27:885–899
Koskakovsky Pond SL, Frost SDW (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21(10):2531–2533
Miller MM, Goto RM, Taylor RL Jr, Zoorob R, Auffray C, Briles RW, Briles WE, Bloom SE (1996) Assignment of Rfp-Y to the chicken major histocompatibility complex/NOR microchromosome and evidence for high-frequency recombination associated with the nucleolar organizer region. Proc Natl Acad Sci USA 93:3958–3962
Murphy K, Travers P, Walport M (2008) Janeway’s immunobiology, 7th edn. Garland Science, New York
Nakagawa S, Burke T (2008) The mask of seniority? A neglected age indicator in house sparrows Passer domesticus. Avian Biol 39:222–225
Nakagawa S, Lee JW, Woodward BK, Hatchwell BJ, Burke T (2008) Differential selection according to the degree of cheating in a status signal. Biol Lett 4:667–669
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Ockendon N, Griffith SC, Burke T (2009) Extrapair paternity in an insular population of house sparrows after the experimental introduction of individuals from the mainland. Behav Ecol 20:305–312
Potts WK, Wakeland EK (1990) Evolution of diversity at the major histocompatibility complex. Trends Ecol Evol 5:181–187
Promerova M, Albrecht T, Bryja J (2009) Extremely high MHC class I variation in a population of a long-distance migrant, the Scarlet Rosefinch (Carpodacus erythrinus). Immunogenetics 61:451–461
Reed KM, Bauer MM, Monson MS, Benoit B, Chaves LD, O’Hare TH, Delany ME (2011) Defining the Turkey MHC: identification of expressed class I- and class IIB-like genes independent of the MHC-B. Immunogenetics 63:753–771
Rodgers JR, Cook RG (2005) MHC class IB molecules bridge innate and acquired immunity. Nat Rev Immunol 5:459–471
Salter RD, Norment AM, Chen BP, Clayberger C, Krensky AM, Littman DR, Parham P (1989) Polymorphism in the α3 domain of HLA-A molecules affects binding to CD8. Nature 338:345–347
Salter RD, Benjamin RJ, Wesley PK, Buxton SE, Garrett TPJ, Clayberger C, Krensky AM, Norment AM, Littman DR, Parham P (1990) A binding-site for the T-cell co-receptor Cd8 on the alpha-3 domain of Hla-A2. Nature 345:41–46
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Habour Laboratory Press, Cold Spring Harbour
Schroeder J, Burke T, Owens I, Nakagawa S (2012) Maternal effects and heritability of annual productivity. J Evol Biol 25:149–156
Schut E, Aguilar JR, Merino S, Magrath MJ, Komdeur J, Westerdahl H (2011) Characterization of MHC-I in the blue tit (Cyanistes caeruleus) reveals low levels of genetic diversity and trans-population evolution across European populations. Immunogenetics 63:531–542
Sepil I, Moghadam HK, Huchard E, Sheldon BC (2012) Characterization and 454 pyrosequencing of Major Histocompatibility Complex class I genes in the great tit reveal complexity in a passerine system. BMC Evol Biol 12:68. doi:10.1186/1471-2148-12-68
Shawar S, Vayas J, Rodgers J, Rich R (1994) Antigen presentation by major histocompatibility class I-B molecules. Annu Rev Immunol 12:839–880
Shiina T, Shimizu S, Hosomichi K, Kohara S, Watanabe S, Hanzawa K, Beck S, Kulski JK, Inoko H (2004) Comparative genomic analysis of two avian (quail and chicken) MHC regions. J Immunol 172:6751–6763
Sibly RM, Witt CC, Wright NA, Venditti C, Jetz W, Brown JH (2012) Energetics, lifestyle, and reproduction in birds. Proc Natl Acad Sci USA 109:10937–10941
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57:758–771
Strand T, Westerdahl H, Hoeglund J, Alatalo RV, Siitari H (2007) The Mhc class II of the Black grouse (Tetrao tetrix) consists of low numbers of B and Y genes with variable diversity and expression. Immunogenetics 59:725–734
Strandh M, Lannefors M, Bonadonna F, Westerdahl H (2011) Characterization of MHC class I and II genes in a subantarctic seabird, the Blue Petrel, Halobaena caerulea (Procellariiformes). Immunogenetics 63:653–666. doi:10.1007/s00251-011-0534-8
Stuglik MT, Radwan J, Babik W (2011) jMHC: software assistant for multilocus genotyping of gene families using next-generation amplicon sequencing. Mol Ecol Resour 11(4):739–742. doi:10.1111/j.1755-0998.2011.02997.x
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121. http://www.kumarlab.net/publications
Westerdahl H (2007) Passerine MHC: genetic variation and disease resistance in the wild. J Ornithol 148:S469–S477
Westerdahl H, Wittzell H, von Schantz T (1999) Polymorphism and transcription of Mhc class I genes in a passerine bird, the great reed warbler. Immunogenetics 49:158–170
Westerdahl H, Wittzell H, von Schantz T, Bensch S (2004) MHC class I typing in a songbird with numerous loci and high polymorphism using motif-specific PCR and DGGE. Heredity 92:534–542
Wittzell H, Madsen T, Westerdahl H, Shine R, von Schantz T (1998) MHC variation in birds and reptiles. Genetica 104:301–309
Wittzell H, Bernot A, Auffray C, Zoorob R (1999) Concerted evolution of two Mhc class II B loci in pheasants and domestic chickens. Mol Biol Evol 16:479–490
Yeager M, Hughes AL (1996) Trans-species polymorphism vs independent evolution of class II MHC sequence motifs. Hum Immunol 47:O689
Zagalska-Neubauer M, Babik W, Stuglik M, Gustafsson L, Cichon M, Radwan J (2010) 454 sequencing reveals extreme complexity of the class II Major Histocompatibility Complex in the collared flycatcher. BMC Evol Biol 10:395. doi:10.1186/1471-2148-10-395
Acknowledgments
We would like to thank Terry Burke for letting us use DNA from the sparrows on Lundy and the Landmark trust and the Lundy Company for letting us work on Lundy. We are grateful to all the staff on Lundy especially Kevin Welsh, Patricia, and Roger Fursdon. Thanks to Ian Cleasby for assistance in the field. We are grateful to Mimi Lannefors and Anna Drews for help in the laboratory and to Emily O’Connor, Kristin Scherman, and Maria Strandh for valuable comments on earlier versions of the manuscript. The project was funded by the Swedish Research Council, VR (621-2206-2876) to Helena Westerdahl and Stiftelsen Lunds Djurskyddsfond to Maria Karlsson.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
239_2013_9575_MOESM1_ESM.pdf
Supplementary Fig. 1. Alignment of house sparrow MHC-I amino acid sequences (Pado-UA), covering the α2 region, retrieved from 454-amplicon sequencing, species specific nomenclature and GenBank accession numbers are used for all sequences (Pado-UA*200-260 (novel from the present study), Pado-UA*324 JN609623, Pado-UA*326 JN609626, Pado-UA*312 JN609636, Pado-UA*309 JN609635, Pado-UA*322 JN609643, Pado-UA*317 JN609640 (found previously and also in the present study) in comparison with the house sparrow transcripts (Pado-UA*230-233, *240-243, this study), great reed warbler (Acar cN3, AJ005503; cN15, AJ005505) and chicken (MHC-B, Gaga, HQ141386; MHC-Y NM_001030675). Amino acid sequences are numbered according to full-length chicken MHC-I. Pado-UA*240-242 have no deletions, Pado-UA*243 has a 3 bp deletion (site 149) and Pado-UA*230-233 have 6 bp deletions (site 246-247). Identity with sequence Pado-UA*240 is indicated with dots, codons corresponding to the PBR according to human HLA (Björkman et al. 1987) with (P); sites subject to negative selection (N) and to positive selection (+) according to HyPhy. HyPhy was run for 38 454-sequences with a 6 bp deletion (with 6 bp deletion), for 20 454-sequences without a 6 bp deletion (without 6 bp deletion) and for the 58 sequences combined (all 454-sequences) (PDF 21 kb)
239_2013_9575_MOESM2_ESM.pdf
Supplementary Fig. 2. Phylogenetic reconstruction of house sparrow (Pado-UA*230-233 and Pado-UA*240-243) and great reed warbler (Acar cN3, AJ005503; cN15, AJ005505) exon 2, 3 and 4 MHC-I nucleotide sequences, with chicken sequences (MHC-B, Gaga, HQ141386; MHC-Y, NM_001030675) as outgroups, using Neighbor-joining (Jukes-Cantor, bootstrap (bt) values based on 1000 replicates). (a) Phylogenetic reconstruction based on nucleotides in exon 2, (b) phylogenetic reconstruction based on nucleotides in exon 3, (c) phylogenetic reconstruction based on nucleotides in exon 4, (d) phylogenetic reconstruction based on synonymous substitutions in exon 2, (e) phylogenetic reconstruction based on synonymous substitutions in exon 3, (f) phylogenetic reconstruction based on synonymous substitutions in exon 4, (g) phylogenetic reconstruction based on nonsynonymous substitutions in exon 2, (h) phylogenetic reconstruction based on nonsynonymous substitutions in exon 3 and (i) phylogenetic reconstruction based on nonsynonymous substitutions in exon 4. The house sparrow sequences with a 6 bp deletion form a significantly supported monophyletic cluster in all trees based on exons 2 and 3 (a bt = 99,b bt = 100,d bt = 77,e bt = 82,g bt = 99,and h bt = 99), but not in trees based on exon 4 (c, f, i) (PDF 18 kb)
239_2013_9575_MOESM3_ESM.jpg
{kind=link}
Supplementary Fig. 3. Inheritance patterns of house sparrow exon 3 sequences (in this paragraph called alleles, with (‘short’) and without (‘long’) the 6bp deletion) in three house sparrow families with 2-4 chicks. Alleles are retrieved from 454-amplicon sequencing, species specific nomenclature and GenBank accession numbers are used for all sequences (Pado-UA*200-260 (novel from the present study), Pado-UA*324 JN609623, Pado-UA*326 JN609626, Pado-UA*312 JN609636, Pado-UA*309 JN609635, Pado-UA*322 JN609643, Pado-UA*317 JN609640 (found previously and also in the present study). Male alleles are indicated with blue, and the two male haplotypes are indicated light and dark blue. Female alleles are indicated with red and the two female haplotypes are pink and red. When alleles are found in both the male and female in-pair haplotypes cannot be inferred and these alleles are non-informative in considering inheritance (indicated green). In clutch 3 the female has three ‘short’ alleles, one allele is red and is inherited in all chicks except in Chick2, Chick2 inherits two pink alleles. This female furthermore has three ‘long’ alleles, one is red and is inherited in all four chicks except in Chick2, Chick2 again inherits two pink alleles. In clutch 3 the male has eight ‘short’ alleles, four are dark blue and they are inherited in all four chicks. This male has six ‘long’ alleles, three are dark blue and are inherited in all four chicks. This male also has four ‘short’ light blue alleles and three ‘long’ light blue alleles, these alleles are not found in any of the four chicks. We find the same pattern of inheritance independent of whether we study inheritance of ‘long’ or ‘short’ alleles, this finding indicates that the ‘long’ and ‘short’ alleles are linked, hence found in the same chromosomal region. Three individuals with 454-read numbers lower than 120 for the ‘short’ alleles were added in this analysis (number of reads from amplification with the ‘short’ primer combination; 64 in Chick2 Clutch3, 65 inChick2 Clutch2 and 92 in the male in Clutch1) (JPG 248 kb)
239_2013_9575_MOESM4_ESM.docx
Supplementary Table 1. Verified MHC-I exon 2-4 cDNA sequences, covering the α1, α2 and α3 regions, in individual 1 and 2 (Ind1 and Ind2), respectively (with a 6 bp deletion, Pado-UA*230-233; without a 6 bp deletion, Pado-UA*240-343) (DOCX 14 kb)
Rights and permissions
About this article
Cite this article
Karlsson, M., Westerdahl, H. Characteristics of MHC Class I Genes in House Sparrows Passer domesticus as Revealed by Long cDNA Transcripts and Amplicon Sequencing. J Mol Evol 77, 8–21 (2013). https://doi.org/10.1007/s00239-013-9575-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9575-y