
Abstract
The great polymorphism observed in the major histocompatibility complex (MHC) genes is thought to be maintained by pathogen-mediated selection possibly combined with MHC-disassortative mating, guided by MHC-determined olfactory cues. Here, we partly characterize the MHC class I and II B of the blue petrel, Halobaena caerulea (Procellariiformes), a bird with significant olfactory abilities that lives under presumably low pathogen burdens in Subantarctica. Blue petrels are long-lived, monogamous birds which suggest the necessity of an accurate mate choice process. The species is ancestral to songbirds (Passeriformes; many MHC loci), although not to gamefowls (Galliformes; few MHC loci). Considering the phylogenetic relationships and the low subantarctic pathogen burden, we expected few rather than many MHC loci in the blue petrel. However, when we analysed partial MHC class I and class II B cDNA and gDNA sequences we found evidence for as many as at least eight MHC class I loci and at least two class II B loci. These class I and II B sequences showed classical MHC characteristics, e.g. high nucleotide diversity, especially in putative peptide-binding regions where signatures of positive selection was detected. Trans-species polymorphism was found between MHC class II B sequences of the blue petrel and those of thin-billed prion, Pachyptila belcheri, two species that diverged ∼25 MYA. The observed MHC allele richness in the blue petrel may well serve as a basis for mate choice, especially since olfactory discrimination of MHC types may be possible in this species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) plays a crucial role in vertebrate adaptive immunity. The genes encoding the MHC class I and II molecules are highly polymorphic and pathogen-mediated selection is generally thought to be a major factor maintaining this polymorphism (Bernatchez and Landry 2003; Doherty and Zinkernagel 1975; Jeffery and Bangham 2000). MHC class I molecules are expressed on all nucleated cells and are involved in presentation of peptides originating from intracellular pathogens (e.g. many viruses), whereas MHC class II molecules only are expressed on immune cells and present peptides derived from extracellular pathogens (e.g. many bacteria). The presentation of non-self peptides on MHC molecules leads to an immune response that ultimately terminates the infection (Janeway et al. 2001).
Mating preferences, for compatibility or “good-genes” (Mays and Hill 2004), guided by MHC-determined olfactory cues has also been suggested to play a role for maintaining the MHC polymorphism (Penn and Potts 1998; Penn 2002; Penn and Potts 1999; Yamazaki et al. 1976). The ability to “sniff out” MHC differences between individuals has been found in various vertebrates including humans (Chaix et al. 2010; Havlicek and Roberts 2009). Mice and rats can distinguish individuals that only differ genetically at the MHC loci (Brown et al. 1987; Yamazaki et al. 1979) and both MHC class I and II loci contribute to personal odours in rats (Brown et al. 1989). In the fish arctic charr, Salvelinus alpinus, and the three-spined stickleback, Gasterosteus aculeatus, MHC class II differences between individuals can be evaluated (Aeschlimann et al. 2003; Olsén et al. 1998). Whether olfactory MHC discrimination is a general feature of considerable evolutionary importance among vertebrates is unclear (Penn and Potts 1998), and for birds this information is by large lacking.
Procellariiform birds (petrels) have a well-developed olfactory system (Bang and Cobb 1968), are long-lived and engaged in lifetime monogamy (Warham 1996), which should make mate choice very important to maximize individual fitness (Bonadonna and Nevitt 2004; Zelano and Edwards 2002). The blue petrel, Halobaena caerulea (Procellariiformes), is a burrow nesting bird with a circumpolar distribution in Antarctic and subantarctic seas, and breeds in dense colonies on small islands close to the Polar Front in the Southern Ocean (Brooke 2004). Daily life of the blue petrel is facilitated by olfaction: food search is odour mediated (Bonadonna et al. 2006) and the blue petrel can identify its underground nest and partner based solely on olfactory cues (Bonadonna et al. 2004; Mardon and Bonadonna 2009; Mardon et al. 2010). In addition, the ability to recognize the partner and avoid self in favour of other individuals in odour choice tests (Mardon and Bonadonna 2009) raises the question whether the blue petrel MHC system could be as important for mate choice/personal recognition as described in some mammals (Penn 2002; Penn and Potts 1999). These characteristics make this species a good model for investigating MHC in birds.
The copy number of MHC genes in birds is highly variable even within orders. The MHC of the galliform red jungle fowl, Gallus gallus, has been shown to be “minimal essential” containing few loci (Kaufman et al. 1999, 1995) whereas the opposite is found in the quail, Coturnix japonica, a bird belonging to the same order (Shiina et al. 2004). Among passerine birds the number of MHC loci is generally higher than for the quail (Balakrishnan et al. 2010; Bollmer et al. 2010; Westerdahl et al. 2000, 2004). The bird order Procellariiformes emerged after Galliformes but before Passeriformes (Hackett et al. 2008) and based on phylogeny we therefore predict the number of MHC loci in the blue petrel to be intermediate to birds in these two orders. Additionally, pathogen pressure and immune investment in birds are lower in colder than in warmer regions, and in marine compared to terrestrial environments (Møller 1998; Piersma 1997). Hence, the pathogen pressure should be comparatively low in the Subantarctics and not selecting as strongly for a multi-loci MHC in the blue petrel as in birds in warmer climates.
Here, we set out to investigate the structural arrangement of the blue petrel MHC in comparison to other birds. This initial characterization is essential for addressing future questions on the role of MHC in mate choice in this species. We analyse both MHC class I and II since odour-mediated discrimination in other species has been shown correlated to either or both MHC classes. More specifically, we examine the number of MHC class I and II B loci, the gene structure and diversity as well as patterns of selection pressure. We base our MHC characterization on both partial cDNA and genomic DNA sequences.
Material and methods
Birds and sampling
A colony of blue petrels, H. caerulea, was studied on Ile Verte, a small island of the Morbihan gulf of Kerguelen, in the southern Indian Ocean (49°51′ S, 70°05′ E), where we yearly followed about 80 nest burrows since 2001. In spite of the fact that the actual population of the island is not estimated, we can consider that all breeding birds in the gulf represent a population of 100,000-200,000 individuals (Weimerskirch et al. 1989). Almost nothing is known about wintering areas of blue petrels. Nevertheless, it has been inferred by diet samples that these Procellariiforms feed in southern Antarctic waters, >1,000 km from the breeding colonies and that blue petrels also foraged on their way back to Iles Kerguelen from these distant foraging grounds (Cherel et al. 2002).
Most birds from the nests we followed are ringed and burrows have been fitted with a closable aperture above the incubating chamber to facilitate capture. Removing birds from the burrow for a brief time does not appear to affect incubation behaviour or the hatchability of the eggs (Bonadonna and Nevitt 2004; Bonadonna et al. 2004). In this study, 25 individuals were included, the individuals were randomly selected and most likely are not members of extended families; hence, the number of individuals should be sufficient for an initial characterization of the blue petrel MHC. Blood samples (0.1–0.5 ml of blood) were collected with syringe from the brachial vein and were kept in Queen’s lysis buffer (0.5 ml; 10 mM Tris–HCl, 10 mM NaCl, 10 mM EDTA, 1% n-lauroylsarcosine, pH 8.0), stored at +4°C until DNA extraction. Additionally, blood (0.1–0.5 ml) was collected for RNA extraction from five individuals in 250 μl K2EDTA (0.2 M) and 500 μl Trizol-LS (Invitrogen, Paisley, UK) was added immediately. The samples were kept at +4°C for approximately 2 months (due to transportation from the remote field site) until RNA extraction.
DNA and cDNA preparations
DNA was extracted from the blood samples, stored in Queen’s buffer, either with standard phenol-chloroform extraction (Sambrook et al. 1989) or with the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The DNA was kept at −20° until used. Total RNA was extracted from the samples collected in Trizol-LS (Invitrogen, Paisley, UK) following the manufacturer’s instructions (carefully collecting only the RNA-containing aqueous phase to avoid DNA contamination) and the integrity of the RNA and the 260/280 ratio was checked. Until cDNA synthesis, the RNA was kept at −80°C. The RNA was reverse transcribed to cDNA using the RETROscript Kit (Applied Biosystems/Ambion, Austin, USA) according to the instructions supplied by the manufacturer.
Sequencing of MHC class I and II B fragments
The main focus in our sequencing efforts was targeting the MHC class I since it has not been described in the Procellariiform order before, whereas the MHC class II B was previously characterized in the thin-billed prion. Pachyptila belcheri (Procellariiformes) (Silva and Edwards 2009). Primers for amplification of MHC class I and class II B genes in the blue petrel were designed (Table 1). A first set of degenerated primers for amplification of 650-bp long MHC class I fragments using cDNA templates, from the middle of exon 2 almost to the end of exon 4, were designed in conserved regions identified in bird MHC sequences available in the GenBank database (Benson et al. 2008). A second primer set for amplification of the variable exon 3 region of class I sequences was designed covering the main part of the exon. Both cDNA and genomic DNA (gDNA) were used as templates. For MHC class II B, a first primer set for amplification of 749-bp long fragments from cDNA, starting at the first base of the coding sequence ending nearly at the end of exon 4, was designed from the full length information of the gene in the closely related thin-billed prion, P. belcheri (Silva and Edwards 2009). Two sets of primers were designed from a number of the 749-bp long sequences resulting from the first class II B amplification, for amplification from cDNA and gDNA of the main part of class II B exon 2 fragments. The MHC fragments were amplified using standard PCR protocols and annealing temperatures specified in Table 1. PCR products were assessed by gel electrophoresis and cloned using the commercially available cloning kits, TOPO TA Cloning Kit with TOPO TA PCR 2.1 vector and One Shot Chemically competent cells (Invitrogen, Paisley, UK) or pGEM-T Easy Vector Systems (Promega, Madison, USA), according to the manufacturers’ instructions. Plasmid clones were transferred to 100 μl of molecular grade H2O in 96-well microtiter plates and immediately lysed at 98°C for 5 min and then frozen at −20°C until analysis. PCR amplifications of the plasmid inserts were performed on the bacterial lysates using pUC/M13 forward and reverse sequence-binding site primers M13F (5′ GTAAAACGACGGCCAG) and M13R (5′ CAGGAAACAGCTATGAC) and standard PCR protocols, followed by assessment of the size and quality of the products by gel electrophoresis. The PCR products were purified and DNA was sequenced using the dideoxy chain-termination method (Sanger et al. 1977). The long (class I, 650 bp; class II B, 749 bp) fragments were sequenced in both directions whereas the class I exon 3 and class II B exon 2 fragments were analysed by single-pass sequencing in the forward direction. Clones containing fragments of expected size were sequenced. From the respective long fragment clonings (class I and II B), 40–50 clones/individual were sequenced for mainly two individuals (Supplementary Table 1). Altogether 313 MHC class I exon 3 clones from 23 individuals (seven out of these individuals were extensively investigated with 20–35 clones/individual) and 92 MHC class II B exon 2 clones from four individuals (maximum 27 clones/individual) were sequenced (Supplementary Table 1). Only presumably functional sequences (without deletions resulting in an incorrect reading frame) verified in two independent PCR reactions were included in further analysis. Before analysing the sequences, the primer sequences were removed. For the data analysis of MHC class I exon 3 and class II B exon 2 sequences, sequence information from the long sequences covering those exons was also included.
Southern blot hybridization
Genomic DNA (8–10 μg) from blue petrel was digested with Pvu II restriction enzyme and then size separated in an agarose gel as described (Westerdahl et al. 1999). The DNA was transferred to a nylon membrane (GE Osmonics labstore, Minnetonka, USA) and two southern blot hybridizations were performed (Westerdahl et al. 1999) using either a blue petrel MHC class I or a MHC class II B probe. The probes were made from purified PCR products of randomly picked clones transformed with a class I exon 3 fragment (primers A21P2–A23H7) or a class II B exon 2 fragment (primers P114–P115). The PCR products were labelled using Amersham Nick Translation Kit (GE Healthcare, Piscataway, USA) according to the manufacturer’s instructions.
Data analysis
The obtained sequences were checked for homology to known MHC genes deposited in GenBank (Benson et al. 2008) and positive sequences were analysed within each MHC class (I or II B) for similarity, divergence and selection. Peptide-binding regions (PBR) and exon boundaries for MHC class I were inferred from chicken and human (Wallny et al. 2006) and for MHC class II from human (Brown et al. 1993).
Apart from the sequences obtained in this study, MHC class I sequences from other bird species, with full coverage of the 606-bp (650 bp including primers) MHC class I fragments amplified in the blue petrel, were downloaded from GenBank and used in a comparative analysis. The species and sequences included were the greylag goose, Anser anser (GenBank accession numbers, Acc nr AM114924, AY387648–AY387652, DQ251182), the mallard, Anas platyrhyncos, (Acc nr AB115241–115246, AF393511, AY294416–AY294419 and AY841881–AY841884), the red jungle fowl, G. gallus (Acc nr AB159063–AB159076, AB178042–AB178045, AB178047, AB178048, AB178050, AB178051, AB178596, AY234769, AY234770, AY821520, GU451331 and NM_001097530) and the great reed warbler, Acrocephalus arundinaceus (Acc nr AJ005503–AJ005509). The sequences were trimmed to the same length as the blue petrel sequences (606 bp) and will from now on be referred to as the “Aves class I dataset”.
Sequences were aligned with ClustalW2 under an IUB cost matrix (Chenna et al. 2003) and bootstrap consensus trees (2,000 replicates) were constructed with the neighbour-joining method in MEGA4 (Felsenstein 1985; Saitou and Nei 1987; Tamura et al. 2007). Nucleotide distances using the Kimura 2-parameter (Kimura 1980) and amino acid p-distances were estimated in MEGA4 (Tamura et al. 2007).
The number of nonsynonymous substitutions per nonsynonymous site (d N) and synonymous substitutions per synonymous site (d S) were estimated from averaging over all sequence pairs. Analyses were conducted using the Nei-Gojobori method in MEGA4 (Nei and Gojobori 1986; Tamura et al. 2007). Standard error estimates were obtained by a bootstrap procedure (1,000 replicates).
Positively selected amino acid sites were estimated in CODEML, a codon-based likelihood programme in the PAML 4 package (Yang 2007), likelihood analyses were conducted under the following random-sites models: M1a (nearly neutral), M2a (positive selection), M7 (β) and M8 (β+ω), where the two latter models should be robust against recombination (Anisimova et al. 2003). If the selection models M2a or M8 fit significantly better than their neutral counter parts, Bayesian statistics integrated in CODEML were used to identify codon sites under positive selection (Yang 2006).
Results
MHC class I
Gene structure
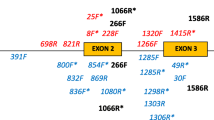
We obtained eight verified unique 606-bp long MHC class I cDNA sequences (partial exon 2–exon 4) from two blue petrel individuals (four per individual) designated by Haca-UA*06L/22L/25L/27L/33L/34L/35L/42L (L, long; Acc nr JF276877–JF276884) according to established nomenclatural rules (Klein et al. 1990) (Supplementary Table 1). In addition, we found another 41 non-verified sequences (in total 16 versus 25 sequences per individual). The overall structure of the 606-bp Haca-UA(L) sequences was similar to the corresponding genes in four other bird orders (Fig. 1). The closest GenBank homolog (89–90% identity) to the eight blue petrel sequences was the Florida sandhill crane, Grus canadensis pratensis, MHC class I mRNA (Acc nr AF033106) which was also the only described gene that clustered with the blue petrel sequences (Supplementary Fig. 1). For the Aves class I dataset (greylag goose, mallard, red jungle fowl and great reed warbler) the MHC class I sequences all clustered according to species (Supplementary Fig. 1). The same pattern was found also when analysing the sequences exon by exon (2–4).

Amino acid alignment of major histocompatibility complex class I translated sequences (Haca-UA*xxL, GenBank accession numbers, Acc nr JF276877–JF276884) from the blue petrel, H. caerulea, five other bird species (Acc nr and species codes are given: Grca Grus canadensis pratensis, Anan Anser anser, Anpl Anas platyrhynchos, Gaga Gallus gallus, Acar Acrocephalus arundinaceus) and human HLA-A (Hosa_NM002116). Peptide-binding regions were inferred (a pocket a, f pocket f, c other peptide contacts) (Wallny et al. 2006). Positively selected amino acid sites (plus sign) identified with model 8 in CODEML, PAML (Yang 2007). Primer sites, A21P2 (F) and A23H7 (R), in exon 3, are underlined
The screening of 168–174-bp MHC class I exon 3 sequences in 23 individuals, resulted in 45 verified sequences, Haca-UA*01–Haca-UA*45 (Acc nr JF276832–JF276876), and 172 non-verified sequences (Supplementary Table 1). The sequences Haca-UA*30 (JF276868) and Haca-UA*39 (JF276872) contained deletions corresponding to a one and two amino acid deletion, respectively (Supplementary Fig. 2).
Alleles and loci
The maximum number of verified genomic exon 3 Haca-UA sequences in one individual was 15 (one of these sequences contained a stop codon), which means that the blue petrel genome should harbour at least eight MHC class I loci. However, the maximum number of transcribed class I sequences (cDNA) in one individual was only nine (Supplementary Table 1) indicating at least five expressed loci. The Southern blot probed with an MHC class I exon 3 fragment showed 11–16 bands per individual (Supplementary Fig. 3). Assuming that each band approximately corresponds to one allele, the southern blot suggests that there are at least eight class I loci in the blue petrel genome.
Divergence and selection
The amino acid distance between the 606-bp MHC class I sequences differed between exons 2, 3 and 4. The distance was significantly larger in the partial exon 2 sequences compared to exons 3 and 4 of the Haca-UA(L) sequences (Fig. 2). This pattern of difference in amino acid distance was also observed in all birds in the Aves class I dataset with exception for the great reed warbler where there was no difference in amino acid distance between exons 2 and 3 (t test, t = 0.1067, p = 0.9168; Fig. 2). The amino acid distance between the Haca-UA(L) sequences was not different from that of any of the other species in exons 2 and 3 but in exon 4 the distance was smaller between the blue petrel sequences than it was between the mallard sequences (t test, t = 2.6101, p = 0.0164; Fig. 2).

Estimates of average evolutionary divergence (amino acid distance) within different exons of major histocompatibility complex class I sequences between five bird species: Haca Halobaena caerulea, blue petrel (Acc nr JF276877–JF276884); Anan Anser anser, greylag goose (Acc nr AM114924, AY387648–AY387652 and DQ251182); Anpl Anas platyrhynchos, mallard (Acc nr AB115241–115246, AF393511, AY294416–AY294419 and AY841881–AY841884); Gaga Gallus gallus, red junglefowl (Acc nr AB159063–AB159076, AB178042–AB178045, AB178047, AB178048, AB178050, AB178051, AB178596, AY234769, AY234770, AY821520, GU451331 and NM_001097530) and Acar Acrocephalus arundinaceus, great reed warbler (Acc nr AJ005503–AJ005509). Sequence information from the middle of exon 2 to the end of exon 4. Analyses were conducted in MEGA4 (Tamura et al. 2007) and standard errors were obtained by bootstrapping (1,000 replicates)
The nucleotide and amino acid distances in the PBR of the 606-bp MHC class I sequences did not differ between the five compared species (the blue petrel and the Aves class I dataset; Table 2). The Haca-UA(L) sequences were less divergent than the greylag goose and the mallard in the non-PBR of exons 2 and 3, as well as exon 4 (Table 2), but not than the red jungle fowl and the great reed warbler.
The overall nucleotide distance between the exon 3 Haca-UA*01–Haca-UA*45 sequences (Acc nr JF276832–JF276876) was found to be 0.076 ± 0.012 (SE) and nucleotide diversity showed higher values in the PBR than in the non-PBR (Table 3).
The nonsynonymous to the synonymous nucleotide substitutions (d N/d S ratio) in the Haca-UA(L) sequences was above one in the PBR of both exons 2 and 3 which indicates that the PBR is under positive selection. For the non-PBR of exons 2 and 3 the d N/d S ratio was lower than one indicating that purifying selection is active in this region (Fig. 3). The estimation of selection on Haca-UA(L) conformed well with the Aves dataset (Fig. 3). Signatures of purifying selection were found for the non-PBR of exon 3 sequences from the screening, whereas the d N was not significantly different from d S in the PBR, although the d N/d S ratio was above one (Table 3).

Average codon-based evolutionary divergence in major histocompatibility complex class I sequences within five bird species: Haca Halobaena caerulea, blue petrel (Acc nr JF276877–JF276884); Anan Anser anser, greylag goose (Acc nr AM114924, AY387648–AY387652 and DQ251182); Anpl Anas platyrhynchos, mallard (Acc nr AB115241–115246, AF393511, AY294416–AY294419 and AY841881–AY841884); Gaga Gallus gallus, red junglefowl (Acc nr AB159063–AB159076, AB178042–AB178045, AB178047, AB178048, AB178050, AB178051, AB178596, AY234769, AY234770, AY821520, GU451331 and NM_001097530) and Acar Acrocephalus arundinaceus, great reed warbler (Acc nr AJ005503–AJ005509). Nonsynonymous (d N) to synonymous (d S) substitution rates. Rates above the broken line signifies sequences under positive selection pressure. Sequence information from the middle of exon 2 to the end of exon 3. Peptide-binding regions (PBR) were inferred (Wallny et al. 2006). Analyses were conducted using the Nei-Gojobori method in MEGA4 (Nei and Gojobori 1986; Tamura et al. 2007)
We also used a codon-based maximum likelihood approach to study positive selection and then found 12 positively selected amino acid sites in exons 2 and 3 for the eight 606-bp Haca-UA(L) sequences with the CODEML programme in the PAML package (Yang 2007) (Table 4; Fig. 1). In the 45 exon 3 Haca-UA sequences, seven positively selected sites were detected with the same method, out of these, four sites were novel compared to the sites identified in the 606-bp sequences (Table 4; Supplementary Fig. 2).
MHC class II B
Gene structure
Two 702-bp (749 bp including primers) MHC class II B cDNA sequences, Haca-DAB*01L and Haca-DAB*06L (partial exon 1–partial exon 4, Acc nr JF276893–JF276894), were verified from two blue petrel individuals. In addition, 32 non-verified sequences with 92–99% nucleotide identity to the verified sequences were found (in total 20 and 12 sequences per individual). The Haca-DAB(L) sequences in the blue petrel (Fig. 4) were very similar (92–93% identity) to the coding sequence of Pabe-DAB1 (Acc nr FJ588549) in the Procellariiform thin-billed prion, P. belcheri, that also served as a template for primer design.

Amino acid alignment of major histocompatibility complex class II B translated sequences (Haca-DAB*xxL, Acc nr JF276893–JF276894) from the blue petrel, H. caerulea, four other bird species (Acc nr and species codes are given: Pabe Pachyptila belcheri, Anpl Anas platyrhynchos, Ancy Anser cygnoides, Gaga Gallus gallus) and human HLA-DRB1 (Hosa_AM493435). Peptide-binding regions inferred from Brown et al. (1993) are shown (c peptide-binding site, p polymorphic peptide-binding site). Primer sites, P141 (F) and P144 (R), in exon 2, are underlined
Eight verified blue petrel 159-bp MHC class II B alleles exon 2 sequences, Haca-DAB*01–Haca-DAB*08 (Acc nr JF276885–JF276892), and another 29 non-verified sequences were obtained from seven birds. The exon 2 Haca-DAB sequences intermixed with the corresponding sequences from the thin-billed prion (Supplementary Fig. 4) and formed a well-supported cluster indicating trans-species evolution of MHC class II B polymorphism between the two (Fig. 5; neighbour-joining of nucleotide sequences, bootstrap value 100%).

Phylogenetic tree of major histocompatibility complex class II B exon 2 (159 bp) nucleotide sequences in the blue petrel, H. caerulea (filled circles Haca-DAB*01–Haca-DAB*08, Acc nr JF276885–JF276892) and the thin-billed prion, P. belcheri (Pabe, Acc nr are given). Two sequences from pheasant, Phasianus colchicus (Phco, Acc nr are given) were used as outgroup. Neighbour-joining bootstrap consensus tree from 2,000 replicates (Felsenstein 1985; Saitou and Nei 1987) constructed in MEGA4 (Tamura et al. 2007). The evolutionary distances were computed using the Kimura 2-parameter method (Kimura 1980)
Alleles and loci
For MHC class II B there were considerably fewer alleles found per individual compared to MHC class I. There were at the most three different verified exon 2 Haca-DAB sequences in one individual (at least two MHC class II B loci). All of them were found in cDNA and were hence expressed. The MHC class II B blot showed three to four bands per individual that hybridized with the exon 2 probe (Supplementary Fig. 3), which suggests that there are at least two class II B loci in the blue petrel genome.
Divergence and selection
The nucleotide distance between the eight exon 2 Haca-DAB*01–Haca-DAB*08 sequences (Acc nr JF276885–JF276892) was estimated to 0.108 ± 0.017 which is slightly, though not significantly, higher than the distance between the class I exon 3 sequences (t test, t = 1.0850, p = 0.2830).
There was support for strong positive selection in the PBR of the exon 2 Haca-DAB sequences (d N/d S ratio = 4.09; Table 3). In the codon-based maximum likelihood analysis, we detected as many as 12 positively selected amino acid sites identified in the exon 2 Haca-DAB sequences with the CODEML-programme in the PAML package (Yang 2007). All of the positively selected sites are within the postulated PBR in the corresponding human HLA-DR1 (Brown et al. 1993).
Discussion
We have characterized the major histocompatibility complex class I and class II B gene structure in a subantarctic Procellariiform seabird, the blue petrel, H. caerulea. We found evidence for at least eight MHC class I loci but only two class II B loci in the blue petrel genome. The high number of class I loci is more similar to the numerous MHC class I loci found in songbirds (Bollmer et al. 2010; Westerdahl et al. 2000, 2004) than to the “minimal essential” MHC found in chicken (Kaufman et al. 1999, 1995). In respects like gene structure, divergence and differential selection pressures in PBR versus non-PBR, the blue petrel MHC conforms well to other bird MHCs.
MHC class I
The 606-bp Haca-UA(L) cDNA sequences had one close GenBank homolog, the Florida sandhill crane MHC class I mRNA (Acc nr AF033106). The blue petrel and the crane belong to Procellariiformes and Gruiformes, orders that separated at least 45 MYA (Sibley and Ahlquist 1990). Neither of the other available long bird MHC class I sequences clustered with the blue petrel sequences but rather by species. The separation between Procellariiformes and Passeriformes is estimated to at least 50 MY and the distance to Galliformes and Anseriformes is at least 64 MY (Sibley and Ahlquist 1990), hence a larger distance than between Procellariiformes and Gruiformes. Divergence times above 45 MY thus reflect species trees rather than gene-trees for the MHC class I among these avian orders. The clustering of sequences exon by exon (2–4) did not change the phylogeny, i.e. there was no evidence for local conservation within the gene structures between species either.
The short distance between the Haca-UA sequences combined with the estimation of loci supports the idea that recent gene duplications and/or gene conversion are common in the avian MHC (Hess and Edwards 2002). On average with few exceptions the blue petrel MHC class I sequences showed the same degree of divergence as their counterparts in other bird species, both regarding the PBR and the non-PBR of the genes.
Curiously, the amino acid distance between the 606-bp MHC class I sequences was significantly larger in the partial exon 2 information (second half of the exon) compared to exons 3 and 4 in all examined bird species except for the great reed warbler. The pattern of a more variable exon 2 compared to exon 3 is also found in the African clawed frog, Xenopus laevis, (Bos and Waldman 2006) which gives some support to this being the ancestral state.
The 3 and 6 bp deletions that were found in the exon 3 sequences Haca-UA*30 (JF276868) and Haca-UA*39 (JF276872) are to date not found at the same positions in other birds, whereas 3 and 6 bp deletions have been reported in transcribed MHC class I exon 3 sequences from the house sparrow Passer domesticus (Bonneaud et al. 2004), Since both Haca-UA*30 and Haca-UA*39 were transcribed and the position of the deletions are within the PBR, the deletions most likely have functional effects in terms of what peptides can bind to the antigen-binding pocket in the encoded MHC molecule. In cattle, Bos taurus, an amino acid deletion detected in the PBR of a MHC gene was predicted to alter the peptide specificity and was shown to be associated with a higher immune response to a vaccine (Sitte et al. 2002).
MHC class II B
The MHC class II B genes in the blue petrel seems to have undergone fewer duplication events than the class I genes since there were considerably fewer alleles found, which was supported also by the southern blot probed with MHC class II B. Perhaps the bacterial diversity in the Antarctics is too low to maintain many class II B loci. Alternatively, there may be loci that our primer sets and probe did not pick up. Compared to what has been found in other birds, especially passerines, the number of class II B loci in the blue petrel was low. However, there are some examples of birds with only one MHC class II B locus found (see comparison in Bollmer et al. 2010). The zebra finch, Taeniopygia guttata, is the complete opposite of the blue petrel considering the MHC loci number, the zebra finch has many MHC class II loci and few class I loci (Balakrishnan et al. 2010). Recently, five MHC class II B loci was found in the little spotted Kiwi, Apteryx owenii, showing that also a species of a basal avian superorder (Paleognathae) can harbour multiple MHC loci (Miller et al. 2011).
The blue petrel and all the prions form two sister clades in a well-supported phylogenetic node where the split probably occurred 24–29.5 MYA (Nunn and Stanley 1998; Penhallurick and Wink 2004). The similarity of the 702-bp Haca-DAB(L) sequences in the blue petrel to the coding sequence of Pabe-DAB1 (Acc nr FJ588549) in the thin-billed prion, P. belcheri, is thus expected phylogenetically even if the divergence time is long. Exon 2 sequences that encode the PBR, which is the most variable part of the gene, from the two sister species clustered together in a phylogenetic tree instead of by species. Hence, there was evidence for trans-species polymorphism between the two species that could either result from their phylogenetic relationship and/or as a result of convergent evolution due to similar selection pressure from pathogens. Trans-species evolution of MHC polymorphism between avian species was reported e.g. among different penguin species, genus Spheniscus, (Kikkawa et al. 2009) and between great reed warbler and Seychelles warbler, Acrocephalus sechellensis (Richardson and Westerdahl 2003).
Signatures of selection in MHC class I and II B
Positive selection acting on the PBR has been found in almost all non-model studies through estimation of nonsynonymous to synonymous nucleotide substitutions (reviewed in Bernatchez and Landry 2003). In the blue petrel we compare sequences in some cases originating from different loci since it was not possible to assign alleles to loci, which is a well-known problem when dealing with the polymorphic MHC region in loci-rich non-model bird species (Westerdahl et al. 2000). We found strong differences in selection pressures between the postulated PBR (positive selection) and non-PBR (negative selection) both for class I and class II B genes, in the range of other avian MHC genes.
Most of the positively selected sites identified in the blue petrel MHC genes are known to be involved in antigen peptide binding in human and/or chicken (Brown et al. 1993; Wallny et al. 2006) which indicates that (1) the structure and function is similar to the MHC in well-known model systems and (2) the genes most likely are transcribed and translated into functional MHC molecules.
MHC and ecology of the blue petrel
Since pathogens presumably are rare among birds in Antarctica (Barbosa and Palacios 2009) the maintenance of so many class I loci in the blue petrel genome may be difficult to explain as an effect of pathogen-mediated selection, which in other systems probably is the main factor keeping MHC polymorphism (Bernatchez and Landry 2003; Jeffery and Bangham 2000). On the other hand, these birds are long-lived so they might encounter quite a few pathogens in a lifetime, even with low environmental pathogen levels. Moreover, the blue petrel is a colonial nester that may reuse underground nests (Bonadonna and Mardon 2010). These factors are associated with increased probability of horizontal pathogen transmission and higher immune responses in other bird species (Møller and Erritzøe 1996; Møller et al. 2003), meaning that pathogen-mediated selection may be stronger for the blue petrel than generally expected in cold climates.
Given the ecology of the blue petrel, where individuals return to their nestling areas to breed with only one partner for life (Brooke 2004), a system for kin recognition and inbreeding avoidance should be important (Penn 2002; Penn and Potts 1999; Zelano and Edwards 2002). If the MHC fills that function in the blue petrel, this may further explain the maintenance of many MHC class I loci. Moreover, the well-developed olfaction as well as the avoidance of self-odour (Mardon and Bonadonna 2009) in the blue petrel, suggests that olfactory MHC discrimination is possible in this species (Zelano and Edwards 2002). Mates with dissimilar MHC alleles could be chosen, thereby kin is avoided and MHC polymorphism is propagated to the next generation (Brown 1997; Jordan and Bruford 1998; Mays and Hill 2004; Tregenza and Wedell 2000). There is some evidence for a MHC-based mate choice in birds (Bonneaud et al. 2006; Freeman-Gallant et al. 2003; Gillingham et al. 2009; Richardson et al. 2005) but also reports on no correlation between MHC and bird mate choice (Ekblom et al. 2004; Westerdahl 2004).
Conclusion
Blue petrels have surprisingly many MHC class I loci, whereas they have fewer MHC class II B loci. Different evolutionary histories for MHC class I and II B with more recent gene duplications or gene conversions for class I than class II B may explain this pattern. Signatures of positive selection in the PBR were detected for both classes of MHC genes. Given the individual odour profiles and olfactory recognition of partner in the blue petrel, it is tempting to assume that the described allele richness serves as a basis for mate choice in this species and that the polymorphism thereby is maintained.
References
Aeschlimann PB, Haeberli MA, Reusch TBH, Boehm T, Milinski M (2003) Female sticklebacks Gasterosteus aculeatus use self-reference to optimize MHC allele number during mate selection. Behav Ecol Sociobiol 54:119–126
Anisimova M, Nielsen R, Yang Z (2003) Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 164:1229–1236
Balakrishnan CN, Ekblom R, Voelker M, Westerdahl H, Godinez R, Kotkiewicz H, Burt DW, Graves T, Griffin DK, Warren WC, Edwards SV (2010) Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biology 8:Article No.: 29
Bang BG, Cobb S (1968) The size of the olfactory bulb in 108 species of birds. Auk 85:55–61
Barbosa A, Palacios MJ (2009) Health of Antarctic birds: a review of their parasites, pathogens and diseases. Polar Biol 32:1095–1115
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2008) GenBank. Nucleic Acids Res 36:D25–D30
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bollmer JL, Dunn PO, Whittingham LA, Wimpee C (2010) Extensive MHC class II B gene duplication in a passerine, the common yellowthroat (Geothlypis trichas). J Hered 101:448–460
Bonadonna F, Mardon J (2010) One house two families: petrel squatters get a sniff of low-cost breeding opportunities. Ethology 116:176–182
Bonadonna F, Nevitt GA (2004) Partner-specific odor recognition in an Antarctic seabird. Science (Washington D C) 306:835
Bonadonna F, Villafane M, Bajzak C, Jouventin P (2004) Recognition of burrow's olfactory signature in blue petrels, Halobaena caerulea: an efficient discrimination mechanism in the dark. Anim Behav 67:893–898
Bonadonna F, Caro S, Jouventin P, Nevitt GA (2006) Evidence that blue petrel, Halobaena caerulea, fledglings can detect and orient to dimethyl sulfide. J Exp Biol 209:2165–2169
Bonneaud C, Sorci G, Morin V, Westerdahl H, Zoorob R, Wittzell H (2004) Diversity of Mhc class I and IIB genes in house sparrows (Passer domesticus). Immunogenetics 55:855–865
Bonneaud C, Chastel O, Federici P, Westerdahl H, Sorci G (2006) Complex MHC-based mate choice in a wild passerine. Proc R Soc Biol Sci B 273:1111–1116
Bos DH, Waldman B (2006) Polymorphism, natural selection, and structural modeling of class Ia MHC in the African clawed frog (Xenopus laevis). Immunogenetics 58:433–442
Brooke M (2004) Albatrosses and petrels across the world. Oxford University Press Inc., New York
Brown JL (1997) A theory of mate choice based on heterozygosity. Behav Ecol 8:60–65
Brown RE, Singh PB, Roser B (1987) The major histocompatibility complex and the chemosensory recognition of individuality in rats. Physiol Behav 40:65–74
Brown RE, Roser B, Singh PB (1989) Class I and class II regions of the major histocompatibility complex both contribute to individual odors in congenic inbred strains of rats. Behav Genet 19:659–674
Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature (London) 364:33–39
Chaix R, Cao C, Donnelly P (2010) Is mate choice in humans MHC-dependent? PLoS Genetics 6:Article No.: e1000184
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Cherel Y, Bocher P, Trouve C, Weimerskirch H (2002) Diet and feeding ecology of blue petrels Halobaena caerulea at Iles Kerguelen, Southern Indian Ocean. Mar Ecol Prog Ser 228:283–299
Doherty PC, Zinkernagel RM (1975) A biological role for the major histo compatibility antigens. Lancet 1:1406–1409
Ekblom R, Saether SA, Grahn M, Fiske P, Kalas JA, Hoglund J (2004) Major histocompatibility complex variation and mate choice in a lekking bird, the great snipe (Gallinago media). Mol Ecol 13:3821–3828
Felsenstein J (1985) Confidence limits on phylogenies and approach using the bootstrap. Evolution 39:783–791
Freeman-Gallant CR, Meguerdichian M, Wheelwright NT, Sollecito SV (2003) Social pairing and female mating fidelity predicted by restriction fragment length polymorphism similarity at the major histocompatibility complex in a songbird. Mol Ecol 12:3077–3083
Gillingham MAF, Richardson DS, Lovlie H, Moynihan A, Worley K, Pizzari T (2009) Cryptic preference for MHC-dissimilar females in male red junglefowl, Gallus gallus. Proc R Soc Biol Sci B 276:1083–1092
Hackett SJ, Kimball RT, Reddy S, Bowie RCK, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han K-L, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Sheldon FH, Steadman DW, Witt CC, Yuri T (2008) A phylogenomic study of birds reveals their evolutionary history. Science (Washington D C) 320:1763–1768
Havlicek J, Roberts SC (2009) MHC-correlated mate choice in humans: a review. Psychoneuroendocrinology 34:497–512
Hess CM, Edwards SV (2002) The evolution of the major histocompatibility complex in birds. Bioscience 52:423–431
Janeway CAJ, Travers P, Walport M, Shlomchik MJ (2001) Immunobiology, the immune system in health and disease. Garland Science, New York
Jeffery KJM, Bangham CRM (2000) Do infectious diseases drive MHC diversity? Microbes Infect 2:1335–1341
Jordan WC, Bruford MW (1998) New perspectives on mate choice and the MHC. Heredity 81:127–133
Kaufman J, Voelk H, Wallny H-J (1995) A "minimal essential MHC" and an "unrecognized MHC": two extremes in selection for polymorphism. Immunol Rev 143:63–88
Kaufman J, Milne S, Goebel TWF, Walker BA, Jacob JP, Auffray C, Zoorob R, Beck S (1999) The chicken B locus is a minimal essential major histocompatibility complex. Nature (London) 401:923–925
Kikkawa EF, Tsuda TT, Sumiyama D, Naruse TK, Fukuda M, Kurita M, Wilson RP, LeMaho Y, Miller GD, Tsuda M, Murata K, Kulski JK, Inoko H (2009) Trans-species polymorphism of the Mhc class II DRB-like gene in banded penguins (genus Spheniscus). Immunogenetics 61:341–352
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219
Mardon J, Bonadonna F (2009) Atypical homing or self-odour avoidance? Blue petrels (Halobaena caerulea) are attracted to their mate’s odour but avoid their own. Behav Ecol Sociobiol 63:537–542
Mardon J, Saunders SM, Anderson MJ, Couchoux C, Bonadonna F (2010) Species, gender, and identity: cracking petrels' sociochemical code. Chem Senses 35:309–321
Mays HL Jr, Hill GE (2004) Choosing mates: good genes versus genes that are a good fit. Trends Ecol Evol 19:554–559
Miller HC, Bowker-Wright G, Kharkrang M, Ramstad K (2011) Characterisation of class II B MHC genes from a ratite bird, the little spotted kiwi (Apteryx owenii). Immunogenetics 63:223–233
Møller AP (1998) Evidence of larger impact of parasites on hosts in the tropics: investment in immune function within and outside the tropics. Oikos 82:265–270
Møller AP, Erritzøe J (1996) Parasite virulence and host immune defense: host immune response is related to nest reuse in birds. Evolution 50:2066–2072
Møller AP, Erritzøe J, Saino N (2003) Seasonal changes in immune response and parasite impact on hosts. Am Nat 161:657–671
Moon DA, Veniamin SM, Parks-Dely JA, Magor KE (2005) The MHC of the duck (Anas platyrhynchos) contains five differentially expressed class I genes. J Immunol 175:6702–6712
Nei M, Gojobori T (1986) Simple methods for estimating the numbers of synonymous and non synonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nunn GB, Stanley SE (1998) Body size effects and rates of cytochrome b evolution in tube-nosed seabirds. Mol Biol Evol 15:1360–1371
Olsén KH, Grahn M, Lohm J, Langefors A (1998) MHC and kin discrimination in juvenile Arctic charr, Salvelinus alpinus (L.). Anim Behav 56:319–327
Penhallurick J, Wink M (2004) Analysis of the taxonomy and nomenclature of the Procellariiformes based on complete nucleotide sequences of the mitochondrial cytochrome b gene. Emu 104:125–147
Penn DJ (2002) The scent of genetic compatibility: sexual selection and the major histocompatibility complex. Ethology 108:1–21
Penn D, Potts W (1998) How do major histocompatibility genes influence odor and mating preferences? Adv Immunol 69:411–435
Penn DJ, Potts WK (1999) The evolution of mating preferences and major histocompatibility complex genes. Am Nat 153:145–164
Piersma T (1997) Do global patterns of habitat use and migration strategies co-evolve with relative investments in immunocompetence due to spatial variation in parasite pressure? Oikos 80:623–631
Richardson DS, Westerdahl H (2003) MHC diversity in two Acrocephalus species: the outbred Great reed warbler and the inbred Seychelles warbler. Mol Ecol 12:3523–3529
Richardson DS, Komdeur J, Burke T, von Schantz T (2005) MHC-based patterns of social and extra-pair mate choice in the Seychelles warbler. Proc R Soc Biol Sci B 272:759–767
Saitou N, Nei M (1987) The neighbor-joining method a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci U S A 74:5463–5467
Shiina T, Shimizu S, Hosomichi K, Kohara S, Watanabe S, Hanzawa K, Beck S, Kulski JK, Inoko H (2004) Comparative genomic analysis of two avian (quail and chicken) MHC regions. J Immunol 172:6751–6763
Sibley CG, Ahlquist JE (1990) Phylogeny and classification of birds: a study in molecular evolution. Yale University Press, New Haven
Silva MC, Edwards SV (2009) Structure and evolution of a new avian MHC class II B gene in a sub-Antarctic seabird, the thin-billed prion (Procellariiformes: Pachyptila belcheri). J Mol Evol 68:279–291
Sitte K, Brinkworth R, East IJ, Jazwinska EC (2002) A single amino acid deletion in the antigen binding site of BoLA-DRB3 is predicted to affect peptide binding. Vet Immunol Immunopathol 85:129–135
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tregenza T, Wedell N (2000) Genetic compatibility, mate choice and patterns of parentage: invited review. Mol Ecol 9:1013–1027
Wallny H-J, Avila D, Hunt LG, Powell TJ, Riegert P, Salomonsen J, Skjodt K, Vainio O, Vilbois F, Wiles MV, Kaufman J (2006) Peptide motifs of the single dominantly expressed class I molecule explain the striking MHC-determined response to Rous sarcoma virus in chickens. Proc Natl Acad Sci U S A 103:1434–1439
Warham J (1996) The behaviour, population biology and physiology of the petrels. Academic Press, London
Weimerskirch H, Zotier R, Jouventin P (1989) The avifauna of the Kerguelen islands Indian ocean. Emu 89:15–29
Westerdahl H (2004) No evidence of an MHC-based female mating preference in great reed warblers. Mol Ecol 13:2465–2470
Westerdahl H, Wittzell H, Von Schantz T (1999) Polymorphism and transcription of Mhc class I genes in a passerine bird, the great reed warbler. Immunogenetics 49:158–170
Westerdahl H, Wittzell H, von Schantz T (2000) Mhc diversity in two passerine birds: no evidence for a minimal essential MHC. Immunogenetics 52:92–100
Westerdahl H, Wittzell H, von Schantz T, Bensch S (2004) MHC class I typing in a songbird with numerous loci and high polymorphism using motif-specific PCR and DGGE. Heredity 92:534–542
Yamazaki K, Boyse EA, Mike V, Thaler HT, Mathieson BJ, Abbott J, Boyse J, Zayas ZA, Thomas L (1976) Control of mating preferences in mice by genes in the major histo compatibility complex. J Exp Med 144:1324–1335
Yamazaki K, Yamaguchi M, Baranoski L, Bard J, Boyse EA, Thomas L (1979) Recognition among mice: evidence from the use of a Y-maze differentially scented by congenic mice of different major histocompatiblity types. J Exp Med 150:755–760
Yang Z (2006) Computational molecular evolution. Oxford University Press, New York
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591
Yang Z, Wong WSW, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zelano B, Edwards SV (2002) An MHC component to kin recognition and mate choice in birds: predictions, progress, and prospects. Am Nat 160:S225–S237
Acknowledgements
This work was supported by the Institut Polaire Francais Paul-Emile Victor (IPEV programme ETHOTAAF no. 354 to FB), and the Agence Nationale de la Recherche Francaise (AMBO ANR-08-BLAN-0117-01 to FB). All aspects of the study were performed according to guidelines established by the IPEV and the CNRS for the ethical treatment of animals and complied with current French regulations. We are grateful to Jerome Mardon, Aurore Malapert and all the field workers who helped through the programme ETHOTAAF over the years. Many thanks to Marie-Pierre Dubois at CEFE, Montpellier and the ecological MHC group in Lund.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Phylogenetic tree of major histocompatibility complex class I exons 2–4 translated sequences (Haca-UA*xxL, GenBank accession numbers, Acc nr JF276877–JF276884) from the blue petrel, Halobaena caerulea, five other bird species (Acc nr and species codes are given: Grca Grus canadensis pratensis, Anan Anser anser, Anpl Anas platyrhynchos, Gaga Gallus gallus and Acar Acrocephalus arundinaceus). Human HLA-A (Hosa_NM002116) was used as outgroup. Neighbour-joining bootstrap consensus tree from 2,000 replicates (Felsenstein 1985; Saitou and Nei 1987) constructed in MEGA4 (Tamura et al. 2007). Filled squares/circles represent sequences from each of two blue petrel individuals (PDF 386 kb)
Supplementary Fig. 2
Amino acid alignment of major histocompatibility complex class I exon 3 translated sequences (Haca-UA*01-Haca-UA*45, Acc nr JF276832–JF276876) from the blue petrel, Halobaena caerulea, five other bird species (Acc nr and species codes are given: Grca Grus canadensis pratensis, Anan Anser anser, Anpl Anas platyrhynchos, Gaga Gallus gallus, Acar Acrocephalus arundinaceus) and human HLA-A (Hosa_NM002116). Peptide-binding regions were inferred (a pocket a, f pocket f, c other peptide contacts, t t-cell contacts) (Wallny et al. 2006). Positively selected amino acid sites (plus sign) identified with model 8 in CODEML, PAML (Yang 2007) (PDF 33 kb)
Supplementary Fig. 3
Southern blots of major histocompatibility complex (MHC) probes hybridized to Pvu II cleaved genomic DNA from four individuals of blue petrels, Halobaena caerulea. Each lane represents an individual (1–4). The left panel shows a MHC class I exon 3 probed blot and the right panel a MHC class II B exon 2 probed blot of the same four individuals (PDF 323 kb)
Supplementary Fig. 4
Amino acid alignment of major histocompatibility complex class II B exon 2 translated sequences (Haca-DAB*01-Haca-DAB*08, Acc nr JF276885–JF276892) from the blue petrel, Halobaena caerulea, and the thin-billed prion, Pachyptila belcheri (Pabe, Acc nr are given). Peptide-binding regions were inferred (c peptide contacts) (Brown et al. 1993). Positively selected amino acid sites (plus sign) identified with model 8 in CODEML, PAML (Yang 2007) (PDF 31 kb)
Supplementary Table 1
Major histocompatibility alleles (class I Haca-UA, class II B Haca-DAB, L long sequences, GenBank Acc nr JF276832–JF276894) identified by sequencing MHC clones from 25 blue petrel, Halobaena caerulea, individuals (1–25; g genomic DNA, c cDNA) (DOC 157 kb)
Rights and permissions
About this article
Cite this article
Strandh, M., Lannefors, M., Bonadonna, F. et al. Characterization of MHC class I and II genes in a subantarctic seabird, the blue petrel, Halobaena caerulea (Procellariiformes). Immunogenetics 63, 653–666 (2011). https://doi.org/10.1007/s00251-011-0534-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-011-0534-8