
Abstract
The purpose of this paper was to optimize an ultra-high performance liquid chromatography (UHPLC) method for the determination and quantification of carotenoid pigments in food products and compare it with a HPLC method in terms of resolution, selectivity, sensitivity, time and reagent saving. Less analysis times, solvent consumption and better peak resolution were achieved when the UHPLC methodology was used. Ultra-high performance liquid chromatography analysis was performed on Zorbax Eclipse Plus C18 column (2.1 × 50 mm; 1.8 μm) using a gradient solvent system and diode array detector. A total of seven carotenoids were separated and quantified in <8.5 min. For all components, the resolution was higher than 1.5 and LOD was below 1.0 μg mL−1. To determine the intraday and reproducibility, assays were conducted in 10 continuous days for method validation achieving recuperation rates ranging between 85 and 100 % and no significant differences among the results obtained in same and different days. The proposed methodology was applied for the determination of the carotenoid contents in honeybee pollen samples, pumpkin and nectarine flesh with satisfactory recovery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carotenoids are a group of natural pigments present in fruits and vegetables imparting them their colours from yellow to red. These pigments are isoprenoids polyenes formed by head-to-tail linkage of C5 isoprene units. Nowadays, the major interest of carotenoids is not only due to their provitamin A activity but also to their antioxidant action by scavenging reactive oxygen species and reducing oxidative stress in the organism [1]. There are many studies showing strong correlations between carotenoid intake and a reduced risk of some diseases, as cancer, atherogenesis, bone calcification, eye degeneration and neuronal damages [2, 3]. In fact, the National Cancer Institute of the United States has recommended an increased intake of food high in carotenoids, thus the relevance of having analytical methods was able to provide more detailed information about the carotenoid composition of foodstuff [4].
For the extraction of carotenoids from the samples, different systems can be used, like liquid–liquid extraction, solid phase extraction or supercritical fluid extraction [5, 6]. AOAC [7] recommends methanol/tetrahydrofuran (THF) (50:50 v/v) for extracting the carotenoids, while other authors use ethyl acetate (100 %) or different mixtures of ethanol/hexane, acetone/ethanol/hexane, ethyl acetate/hexane or acetone/hexane or acetone [8, 9].
Carotenoid analysis in food products may be carried out by different methods: HPLC, spectrophotometry or colour evaluation [10]. Among them, chromatographic separations have major relevance due to the feasibility of this methodology to separate a great variety of these pigments isomers. High performance liquid chromatography analysis of carotenoids is usually done with C18 or C30 RP columns, operated with isocratic or gradient elution with a wide variety of mixtures of different organic solvents as mobile phases, using UV–vis (λ≈450 nm) or diode array detector (DAD) or MS detection [8, 11]. Heating the column is sometimes used to improve pigment separation as well as to standardize the separation conditions [10]. By using HPLC, an average of 20–30 min is needed to obtain a complete elution of a group of carotenoid pigments [12].
Different strategies have been achieved in order to reduce the analysis time in chromatographic-based methodologies. González-Gómez et al. [13] proposed the application of multivariate analysis to chromatographic data to resolve coeluted peaks of carotenoid pigments in vegetable matrices without increasing analysis time. On the other hand, the use of chromatographic systems operating at very high pressure (UHPLC) has gained in importance. The use of this very high pressure together with 1.7-μm-particle size columns allows saving on time and solvents consumption, without altering or even improving sensitivity and peak resolution [14–16].
Thus, the aim of this research was to establish a new UHPLC methodology to analyze carotenoid pigments from complex food-matrix samples. In general terms, adapting the existing HPLC methods to UHPLC requires modifications in chromatographic conditions and sample preparation in order to exploit all the advantages of this new methodology. In addition, in order to evaluate the advantages of the UHPLC methodology, we have compared the performance of the proposed UHPLC method with the existing HPLC application. Since this research work was focused on the UHPLC method development, a previously published carotenoid extractive procedure was employed for all pigment extractions [9].
Thus, the UHPLC methodology proposed in this manuscript will allow carotenoid pigments separation and quantification without loosing chromatographic resolution and saving considerable amount of solvent and time. We have included all statistical analysis of method validation in order to prove the confidence of the proposed methodology.
Materials and methods
Food product samples
In order to validate the proposed methodology with different food matrices, the studies performed in this research were conducted with three different food samples: honeybee-collected pollens, pumpkins and nectarines fruit. All samples were purchased in local markets. Three independent replicas were analyzed for each food sample (n = 3). Carotenoid pigments were extracted accordingly and carotenoid dry extracts were stored at −80 °C until analysis.
Chemical standards and reagents
Capsantine (CAP), α- and β-carotene (α-CAR and β-CAR), β-cryptoxanthin (CRYP), lycopene (LYC), lutein (LUT) and zeaxantin (ZEA) were purchased from Extrasynthese, Lyon (France). All standards were in their trans configuration and their purities were at least at 98 % according to supplier information. All solvents used for standards and samples preparation and for chromatography (acetonitrile, ethyl acetate, methanol) were supplied from Fisher Spain, Madrid (Spain). Ultrapure water was obtained by a Mili-Q Plus water purification system (Millipore).
Standard stock solutions of 100 μg/mL were prepared in chloroform containing 0.1 % BHT in order to increase solution stability. Solutions were stored in the dark at −20 °C. Standard working solutions were prepared from the stock solution by the appropriate dilution in acetonitrile/methanol/ethyl acetate (60:20:20).
HPLC determination
Samples were analyzed using a High Performance Liquid Chromatographer Agilent 1200 (Agilent Technologies, CA, USA) with a Lichrosorb RP-18 column 4.6 × 200 mm (10 μm particle size) coupled to a DAD detector (measuring at 460 nm). The gradient mobile phase was a mix of acetonitrile/methanol 85:15 (solvent A) and acetonitrile/methanol/ethyl acetate 60:20:20 (solvent B) containing 0.1 % BHT (butylated hydroxytoluene) and 0.05 % TEA (triethanolamine) according to the timetable shown in Table 1. The flow rate applied was 1 mL/min and analysis was performed at room temperature and column temperature was maintained at 28ºC using a column oven.
UHPLC determination
Samples were analyzed using an Ultra High Performance Liquid Chromatographer Agilent 1290 (Agilent Technologies, CA, USA) with a Zorbax Eclipse Plus C18 column 2.1 × 50 mm (1.8 μm particle size) coupled to a DAD detector (measuring at 460 nm). The gradient mobile phase was a mix of acetonitrile/methanol 85:15 (solvent A) and acetonitrile/methanol/ethyl acetate 60:20:20 (solvent B) containing 0.1 % BHT and 0.05 % TEA according to the timetable shown in Table 1. Flow rate was 0.3 mL/min and column temperature was maintained at 28 °C using a column oven.
In both cases, peak identification was achieved by the individual injection of each pigment standard.
Carotenoid extraction
The methodology to extract the carotenoid pigments was based on the procedure established by Mínguez-Mosquera and Hornero-Méndez [9], slightly modified. Briefly, the extraction method was: 5 g of food product were extracted four times with 25 mL of acetone in 100 mL volumetric flask and shaken for 30 min. The acetone extracts were transferred to a separation funnel and 30 mL of hexane was added followed by 20 mL of purified water. After a 10 min of shacking, the organic phase was separated in a beaker for vacuum evaporation. The extraction process was carried out in the absence of light in order to prevent pigment degradation. Before analysis, the carotenoid extracts were solved in 2 mL of acetonitrile/methanol/ethyl acetate (60:20:20) containing 0.1 % of BHT and 0.05 % of TEA (chromatographic mobile phase).
Quantification and data analysis
To quantify the concentration of the carotenoid compounds, different regression lines were built for each carotenoid standard. In all cases, the regression lines were constructed injecting five different concentration levels: 0.20, 0.80, 1.50, 2.00, 3.00 μg/mL for all the studied compounds and using both chromatographic methodologies (HPLC and UHPLC). Table 2 summarizes the parameters of each analytical curve for the studied carotenoid compounds. Both procedures were compared in terms of chromatographic peak resolution, repeatability and reproducibility of retention time and peak area and sensitivity. In addition, the concentrations of carotenoids, quantified using the different chromatographic methods, in a significant set of samples, were also compared.
For each carotenoid, the detection limit value (LOD) expressed as μg/mL was calculated following the Long and Winefordner [17] criteria, and the limit of quantification (LOQ) expressed as μg/mL, according to the IUPAC norm; linearity and analytical sensitivity were expressed as mL/μg according to Cuadros [18].
Interday repeatability of retention times and of peak areas was calculated from the repetition of ten injections of a mix of pure standards performed on the same day, and method reproducibility was assessed from the repetition of ten injections of a mix of pure standards performed on different days spread evenly over 10 days. A variance statistical test was applied to evaluate the differences between the results obtained within the day and different days.
Results and discussion
The chromatographic separation was applied to determine carotenoid pigments in three different food matrices: honeybee-collected pollen, pumpkin and nectarine. The selections of these food matrices have different carotenoid pigments and diverse relative abundances. Bee pollen is an apicultural product, which may be used for its nutritional value in the human diet [19]. Honeybee-collected pollen contains significant amounts of polyphenols and carotenoid pigments. Due to the chemical composition of honeybee-collected pollen, it can be considered as a potential source of energy and proteins for human consumption [20]. Pumpkins, which are the fruits of different species of the genus Cucurbita, are cultivated worldwide for their pulp and seeds for human nutrition, either for direct consumption or for preparation of other foods such as purées. Pumpkin pulp has large amounts of carotenoids, which are pigments that derive from isoprene and that give flowers, leaves and fruits a colouration that ranges from yellow to red [21]. Nectarine fruit is rich in ascorbic acid (vitamin C), carotenoids (provitamin A) and phenolic compounds that are good sources of antioxidants [22].
Analytical parameters of the chromatographic methodologies
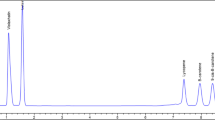
According to the chromatographic conditions described above, a mixture of carotenoid standards was analyzed by means of the HPLC and UHPLC methodologies. The chromatograms obtained are plotted in Fig. 1. In both methodologies, adequate resolution was obtained for CAP, LUT, ZEA, CRYP and LYC, however in the case of α- and β-carotene isomers, the HPLC methodology was not able to separate them, while the UHPLC provided satisfactory resolution (Rs = 2.35 min) (Fig. 1). In addition, the order of elution of carotenoids was not always the same between the two systems: ZEA was eluted before LUT in UHPLC but after LUT in HPLC. As specified above, different silanol activities can modify retention and so the order of elution [23].

Chromatogram belonging to elution of the CAP, LUT, ZEA, CRYP, LYC, α-CAR and β-CAR by the optimized HPLC (a) and UHPLC (b) methodologies
Under the selected conditions, regression lines were obtained by preparing standard samples of the mixture of the seven carotenoids in triplicate, with increasing concentrations of each analyte as it was explained above.
The main figures of merit (quality parameters of the proposed methodologies) were calculated as it was mentioned above. For both models, regression coefficients and linearity values were acceptable for an analytical method (R > 0.99 in most cases and linearity (%) >95 % in most cases). In addition, the LOD and LOQ values reported for the different compounds and methods ensure enough sensitivity for the determination and quantification of these compounds in food material. In addition, no significant differences were observed between the LOD and LOQ values calculated from the HPLC and UHPLC methodologies. The standard error of the regression, calculated by Espinosa-Mansilla et al. [24], indicated that using the UHPLC methodology reported lower error values.
The precision of the method was determined by calculating the intraday repeatability and the interday precision, by injection of ten replicates of standard sample within the day (intraday repeatability) and within 10 days (interday precision). The results are summarized in Table 3a and b (for the HPLC and UHPLC methodologies, respectively) and were evaluated in terms of repetitively of standard sample quantification, retention time of the eluted peaks and resolution values between the chromatographic peaks. For both models, the recovery values of the quantified concentrations ranged between 85 and 100 %. A Fisher statistical test was performed in order to evaluate the interday and intraday precision. The comparison between the quantified concentrations, retention times and peaks resolutions was statistically indistinguishable (Fcal < Ftab), regardless of the fact that the analyses were performed on the same or different days.
Alternative UHPLC procedures can be found in the literature [16, 25] for the analysis of carotenoids in different complex matrices. In the first research work [16], a UPLC methodology is proposed by the analysis of carotenoids, retinol and tocopherols in bovine plasma and milk. In this methodology, a 46-min chromatographic run time are needed to elute 26 peaks. On the other hand, Granado–Lorencio’s work [25] employs 4.5 min to elute nine carotenoids in human serum; however in their methodology, the isomers LUT and ZEA coeluted in the same chromatographic peak.
Analysis of carotenoids in food matrices by the UHPLC method
The proposed UHPLC method was applied to determine the concentration of carotenoid pigments in different carotene-rich food matrices with satisfactory results. Peaks identification in the food samples was determined by comparing their chromatographic retention time and the UV–vis spectra obtained in the chromatographic peak with those from standards. On the other hand, the group of known peaks obtained in the chromatogram of honeybee collected pollen could be xanthophyll esters, and their identification could be achieved by saponification. Figure 2 represents the chromatograms for nectarine, pumpkin and honeybee collected pollen. The UHPLC methodology allowed the separation of the different carotenoids peaks from the matrix background, keeping the resolutions achieved during the method validation. Butylated hydroxytoluene as an antioxidant was added to extraction solvents and mobile phase in order to prevent significant degradation of carotenoids during sample preparation and HPLC analysis, because carotenoids tend to oxidize to colourless degradation products by oxygen and light [26, 27].

UHPLC chromatogram of nectarine, pumpkin and honeybee collected pollen samples
The quantification of the identified peaks for each food matrices is summarized in Table 4. Although the results are comparable, in general terms, with those published by other authors for same food matrices using different analytical methodologies [28–30], the difference in carotenoid content between values reported in our research and the cited references could be attributed to the great variability existing in fruit samples due to ripening stage or fruit cultivar.
Conclusions
In conclusion, we describe here a new gradient method for the UHPLC simultaneous determination of carotenoid contents in food products. The proposed UHPLC method is simple, and a short time, about 10 min, is necessary for elution of the compounds and equilibration of the column. A very good chromatographic resolution was obtained for the seven analytes. The new optimized method might be useful in routine assessment because it saves solvents and chromatographic time.
References
Rao AV, Honglei S (2002) Nutr Res 22:1125–1131
Cantuti-Castelvetri I, Shukitt-Hale B, Joseph JA (2000) Int J Dev Neurosci 18:367–381
Yamaguchi M, Uchiyama S (2003) Biol Pharm Bull 26:1188–1191
Krinsky N, Johnson EJ (2005) Mol Aspects Med 26:459–516
Kozukue N, Friedman M (2003) J Sci Food Agric 83:195–200
Rozzi NL, Singh RK, Vierling RA, Watkins BA (2002) J Agric Food Chem 50:2638–2643
AOAC (Association of official analytical chemists) (1993) AOAC, manual on policies and procedures. AOAC, Arlington
Lin CH, Chen BH (2003) J Chromatogr A 1012:103–109
Mínguez-Mosquera MI, Hornero-Méndez D (1993) J Agric Food Chem 41:1616–1620
Schoefs B (2002) Trends Food Sci Technol 13:361–371
Burns J, Fraser PD, Bramley PM (2003) Phytochemistry 62:939–947
Eder R (2000) In: Nollet LML (ed) Food analysis by HPLC, 2nd edn. Marcel Dekker, New York
González-Gómez D, Lozano M, Fernández-León AM, Fernández-León MF, Cañada-Cañada F (2012) Czech J. Food Sci. 30:358–363
Acquity UPLC columns (2009) Waters corporation, Milford, MA. http://www.waters.com. Accessed 27 Jul 2009
Guillarme D, Nguyen DT-T, Rudaz S, Veuthey JL (2007) J Chromatogr A 1149:20–29
Chauveau-Duriot B, Doreau M, Noziere P, Graulet B (2010) Anal Bioanal Chem 397:777–790
Long GL, Winefordner JD (1983) Anal Chem 55:712–724
Cuadros L, García Campaña AM, Jiménez C, Román Ceba M (1993) Anal Lett 26:1243–1258
Sarmento TM, Amorim C, da Silva AC, Barbosa-Filho JM, Sarmento EM, Magalhaes B, Ribeiro FA (2006) J Food Compos Anal 19:507–511
Kroyer G, Hegedus N (2001) Innov Food Sci Emerg Technol 2:171–174
Oliver J, Palou A (2000) J Chromatogr A 881:543–555
Tomás-Barberán FA, Gil MI, Cremin P, Waterhouse AL, Hess-Pierce B, Kader AA (2001) J Agric Food Chem 49:4748–4760
Sander LC, Sharpless KE, Pursch M (2000) J Chromatogr A 880:189–202
Espinosa-Mansilla A, Muñoz de la Peña A, González-Gómez D (2005) The Chemical Educator 10:337–345
Granado-Lorencio F, Herrero-Barbudo C, Blanco-Navarro I, Pérez-Sacristán B (2010) Anal Bioanal Chem 397:1389–1393
Talwar D, Ha TKK, Cooney J, Brownlee C (1998) St Jo’Reilly D. Clinical Chimica Acta 270:85–100
Gueguen S, Herbeth B, Siest G, Leroy P (2002) J Chromatogr Sci 40:69–76
Gil MI, Tomás-Barberán FA, Hess-Pierce B, Kader AA (2002) J Agric Food Chem 50:4976–4982
de Carvalho LMJ, Gomes PB, Godoy RLDO, Pacheco S, do Monte PHF, de Carvalho JLV, Nutti MR, Neves ACL, Vieira ACRA, Ramos SRR (2012) Food Res Int 47:337–340
Domínguez-Valhondo D, Bohoyo-Gil D, Hernández T, Gonzalez-Gomez D (2011) Int J Food Sci Technol 46:2204–2211
Acknowledgments
Authors thank Junta de Extremadura of Spain and FEDER for financial support (Project GR10006). D.B.G. and D.G.G. thank Instituto Nacional de Investigación and Tecnología Agraria and Alimentaria (INIA) for their research contracts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bohoyo-Gil, D., Dominguez-Valhondo, D., García-Parra, J.J. et al. UHPLC as a suitable methodology for the analysis of carotenoids in food matrix. Eur Food Res Technol 235, 1055–1061 (2012). https://doi.org/10.1007/s00217-012-1838-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-012-1838-0