
Abstract
Screening, isolation and in vitro or in vivo assays have been used for characterisation of bioactive peptides derived from food proteins. Bioinformatic computational methods as quantitative structure–activity relationship (QSAR) and computer-predicted (in silico) proteolysis have been complementary to experimentally work. Recent developments in molecular characterisation and bioinformatics have further made it possible to “dock” small molecules (i.e. ligands) towards proteins and “score” their potential binding. Thus, methods like docking and virtual screening are becoming widely used in drug development, but to our knowledge have found limited use in food science. Angiotensin converting enzyme (ACE) inhibitory dipeptides were therefore docked towards a protein target. A significant relationship was found between results from computational docking and experimental values for inhibition (n=58, R 2=0.28, p<0.001). Docking and virtual screening were found feasible to identify promising bioactive peptide structures and could provide molecular understanding but does not replace the need for experimental verification and analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteins provide essential amino acids and energy required for growth and maintenance of the body and give food physical and functional properties. Peptides and free amino acids in foods are mainly derived from proteolysis of proteins during processing, ripening or eventually by gastrointestinal digestion. Proteolysis is important for development of product quality in, e.g., ripened cheese [1], but recent research also point out that some peptides liberated during proteolysis of food proteins may have physiological activity (bioactivity) [2]. A well-known physiological effect of peptides is taste. Bitter- and/or umami-tasting peptides have been isolated from foods and protein hydrolysates [3]. Some peptides have also shown health-related bioactivity related to the cardiovascular (e.g. angiotensin-I-converting enzyme (ACE) inhibitory peptides and antitrombotic peptides), nervous (e.g. opioid peptides), immune (e.g. immunomodulating), host defence (e.g. antimicrobial peptides), cognitive (e.g. prolyl oligopeptidase (POP) inhibitory peptides) and nutritional system (e.g. casein phosphopeptides) [4]. Bioactive peptides are therefore used as active ingredient in several food products with claimed health effects.
An increasing amount of experimental research on bioactive peptides derived from food proteins are reported in literature. Both in vitro and in vivo assays have been developed to determine bioactivity of isolated peptide fractions from foods or protein hydrolysates. Bioactive peptides have often been synthesised to make more extensive biochemical and biological characterisation possible. Screening studies of foods and process optimisation for efficient production of bioactive peptides have been explored. It has gained insight into active peptide structures and development of novel foods and ingredients [5]. Complementary to experimentally work, have been computational methods to predict and understand relationship between peptide structure, bioactivity and their formation during proteolysis [6]. Databases of food-derived bioactive peptides are useful in that respect [7] and have been used in combination with screening food protein sequences for possible release of bioactive peptides, in silico (computer-predicted) proteolysis and quantitative structure–activity relationship (QSAR) modelling [8–11].
Finding chemical structures with feasible physiological activities is an area driven by medical and pharmaceutical research through drug discovery. Drug discovery is to a large extent based on experimentally screening potentially active compounds for bioactivity with evaluation and modifications of promising lead candidates. Recently, as the information on molecular characteristics and models of binding sites in physiological targets are becoming more available [12, 13], computational approaches that “dock” small molecules into the structures of macromolecular targets and “score” their potential complementarity are becoming widely used in hit identification and lead optimisation [14]. Thus, lead candidates can be found using a docking algorithm that tries to identify the optimal binding model of a small molecule (i.e. ligand) to the active site of a macromolecular target (e.g. active site in an enzyme). Scoring results from computational ligand–protein docking can then be explored for virtual screening. Virtual screening involves databases with relevant molecular structures that are docked into protein targets. Respective scoring results are then used for identification of structures with potential binding and physiological activity, which can be further evaluated experimentally. Development of drugs such as HIV protease inhibitors have been heavily influenced by structure-based design using computational methods and modelling [15, 16]. However, there remain significant challenges in application of such methods and the relationship between computational results and biochemical or biological activity is indeed not straightforward [14, 17].
Virtual screening and ligand–protein docking is a relative unexploited area in the field of bioactive peptides from foods and could contribute to increased insight into molecular mechanisms. It could also be a complementary method for identification of bioactive peptide sequences. ACE inhibitory dipeptides previously used in QSAR modelling were therefore docked towards relevant protein targets. Computational results were compared to experimental values to explore if docking and virtual screening have the potential to identify bioactivity to peptides derived from food proteins.
Materials and methods
Bioactive peptides and target proteins
The protein structure complex of the anti-hypertensive drug captopril with angiotensin-I-converting enzyme (PDB ID: 1UZF) was obtained from the Protein Data Bank (http://www.pdb.org). Data set of dipeptides with experimentally determined values for inhibition of ACE (Table 1) were originally from Cushman et al. [18]. It has been widely used by Hellberg et al. [19], among others, for evaluation of peptide QSAR methods. Observed inhibitory activity of dipeptides were expressed as peptide concentration in molar needed to inhibit 50% of enzyme activity (IC50) and transformed to the logarithmic value of the inverse (log 1/IC50).
Docking
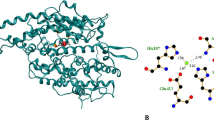
The molecular structure of dipeptides was modelled using the polypeptide builder function in ArgusLab 4.0.1. (Planaria Software LLC, Seattle, WA, http://www.arguslab.com) and saved in the MOL format. Explicit hydrogens in the peptide structures were also assigned by ArgusLab. Protein structures from the Protein Data Bank were saved in their pdb format. Ligand–protein docking was performed with the molecular docking algorithm MolDock [20] using the Molegro Virtual Docker version 1.1.1 software (Molegro ApS, Aarhus, Denmark, http://www.molegro.com) according to instructions. Peptides (ligands) modelled in Arguslab and protein structure complexes were imported into the docking program assigning bonds, hybridisation and explicit hydrogens if missing and always charges and flexible torsions with the Molegro Virtual Docker software. The molecular structure of imported ligands was manually checked before docking and corrected in those cases were it had failed. Water molecules with the protein structures were excluded from the docking experiments, but metal or salt ions were retained. Binding sites were restricted within a 15×15×15 Å3 cube centred at the observed binding of captopril in protein complex PDB ID: 1UZF (Fig. 1).

Protein structure of angiotensin converting enzyme PDB ID: 1UZF with binding site restricted within a 15×15×15 Å3 cube (indicated by green colour) centred at the observed bound captopril molecule
Due to the stochastic nature of the ligand–protein docking search algorithm, 10 runs were conducted and 10 docking solutions (pose) were retained for each ligand. The interaction energy between the pose with highest ranking Moldock score and the protein were compared with experimental values for enzyme inhibition expressed as log (1/IC50). The coefficient of determination (R 2) between predicted ligand–protein interaction and observed enzyme inhibition was determined with corresponding p-values from statistical testing using the Minitab Release 14 Statistical Software (Minitab Ltd., State College, PA, USA).
Results and discussion
The data set of ACE inhibitory dipeptides (Table 1) has also been used for QSAR modelling. It was therefore intentionally chosen, so that the two bioinformatic approaches, i.e. docking and QSAR modelling, could be compared for the ability to identify bioactive peptide sequences. There are several available protein structures for ACE in the Protein Data Bank (http://www.pdb.org). In this study, a protein structure complex without specific mutations and with a bound captopril molecule was chosen. Water molecules are often omitted from comparable docking experiments [20] and were therefore also omitted here. Ions were included, since they are an integral component of many enzymes and are essential to their catalytic function and structural stability [21]. For instance, zinc and chloride ions are involved in the catalytic activity of ACE [22].
Docking of peptide sequences was performed using the MolDock algorithm [20] and relationship between observed enzyme inhibition and the estimated interaction energy between the poses and the proteins was evaluated (Fig. 2). QSAR modelling of the ACE-inhibitory dipeptide data using different amino acid descriptors set have produced somewhat stronger relationship between predicted and measured values (R 2 usually in the area 0.6–0.7) than found in this docking experiment. However, QSAR models are obtained using regression analysis of chemical descriptors and observed values of bioactivity. Whereas docking does not, in a similar direct way, take into account measurements of observed values of bioactivity in the modelling. It is therefore not unexpected that QSAR modelling provides a better relationship than docking for this data set.

A significant relationship was found (n=58, R 2=0.28, p<0.001) between observed inhibition of angiotensin converting enzyme inhibitory dipeptides (Table 1) versus estimated interaction energy between poses and the protein
Even though docking programs and scoring methods have been found fairly able to reproduce the experimental observed binding model and differentiate active ligands from decoys, a major criticism has been their inability to distinguish, differentiate and quantitative the sometimes subtle differences that can change ligand affinity from highly potent to inactive [17]. The modeller must also take great care in expressing correct structural characteristics of ligands and proteins to resemble the given biochemical conditions. Our reported docking study does also reflect this, since a large variation in predicting ability was found. However, the predictive activity seemed in the case of ACE inhibitory dipeptides to be close to what can be obtained using QSAR modelling. Both QSAR modelling and docking should therefore be feasible methods to find promising peptide structures for bioactivity, but does not replace the need for further experimental verification and analysis. Suggestions of promising peptide structures for bioactivity from modelling have to be tried out experimentally and compared with other modified structures to identify highly potent bioactive peptides. However, instead of going directly to large-scale experimental screening to identify novel neutraceuticals, a more structured approach would be molecular modelling and docking to select promising candidates and then assays to measure the activity of selected candidates from docking followed by QSAR modelling to guide further molecular optimisation.
Even though docking does not in any way replace the need for in vitro and in vivo testing, it could contribute to a molecular understanding of bioactivity. That alone, legitimate that it should be more extensively explored in food chemistry to understand how biological active compounds in foods can interact with macromolecular receptor.
References
Sousa MJ, Ardö Y, McSweeney PLH (2001) Int Dairy J 11:327–345. DOI: 10.1016/S0958-6946(01)00062-0
Gobbetti M, Stepaniak L, De Angelis M, Corsetti A, Di Cagno R (2002) Crit Rev Food Sci 42:223–239.
RoudotAlgaron F (1996) Lait 76:313–348
Silva SV, Malcata FX (2005) Int Dairy J 15:1–15. DOI: 10.1016/j.idairyj.2004.04.009
Korhonen H, Pihlanto A (2006) Int Dairy J, in press. DOI: 10.1016/j.idairyj.2005.10.012
Pripp AH, Isaksson T, Stepaniak L, Sørhaug T, Ardö Y (2005) Trends Food Sci Technol 16:484–494. DOI: 10.1016/j.tifs.2005.07.003
Dziuba J, Iwaniak A (2005) Database of protein and bioactive peptide sequences. In: Mine Y, Shahidi F (eds) Neutraceutical proteins and peptides in health and disease. Taylor & Francis, Boca Raton, FL
Dziuba J, Iwaniak A, Minkiewicz P (2003) Polimery 48:50–53
Dzuiba J, Niklewicz M, Iwaniak A, Darewicz M, Minkiewicz P (2004) Acta Aliment Hung 33:227–235
Vermeirssen V, van der Bent A, Van Camp J, van Amerongen A, Verstraete W (2004) Biochimie 86:231–239. DOI 10.1016/j.biochi.2004.01.003
Pripp AH (2005) Eur Food Res Technol 211:712–716. DOI: 10.1007/s00217-005-0083-1
Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J (2000) Nature Struct Biol 7:957–959
Westbrook J, Feng Z, Chen L, Yang H, Bergman HM (2003) Nucleic Acid Res 31:489–491
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Nat Rev Drug Discov 3:935–949
Sangma C, Chuakheaw D, Jongkon N, Saenbandit K, Nunrium P, Uthayopas P, Hannongbua S (2005) Comb Chem High Throughput Screen 8:417–429
Kroeger Smith MB, Hose BM, Hawkins A, Lipchock J, Farnsworth DW, Rizzo RC, Tirado-Rives J, Arnold E, Zhang W, Hughes SH, Jorgensen WL, Michejda CJ, Smith RH Jr (2003) J Med Chem 46:1940–1947. DOI: 10.1021/jm020271f
Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Sanger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2005) J Med Chem ASAP article DOI: 10.1021/jm050362n
Cushman DW, Cheung HS, Sabo EF, Ondetti MA (1981) Angiotensin converting enzyme inhibitors: evolution of a new class of antihypertensive drugs. In: Horovitz (ed) Angiotensin converting enzyme inhibitors. Mechanisms of action and clinical implications. Urban & Schwarzenberg, Baltimore, pp 3–25
Hellberg S, Eriksson L, Johsson J, Lindgren F, Sjöström M, Skagerberg B, Wold S, Andrews P (1989) Int J Peptide Res 37:414–424
Thomsen R, Christensen MH (2006) J Med Chem 49:3315–3321. DOI: 10.1021/jm051197e
Creighton TE (1993) Proteins: structures and molecular properties 2nd ed. WH Freeman, New York
Sturrock ED, Natesh R, van Rooyen JM, Acharya KR (2004) Cell Mol Life Sci 61:2677–2686. DOI: 10.1007/s00018-004-4239-0
Pripp AH, Isaksson T, Stepaniak L, Sørhaug T (2004) Eur Food Res Technol 219:579–583
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pripp, A.H. Docking and virtual screening of ACE inhibitory dipeptides. Eur Food Res Technol 225, 589–592 (2007). https://doi.org/10.1007/s00217-006-0450-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-006-0450-6