
Abstract
Presently, there is a growing interest in the development of lateral flow devices for nucleic acid analysis that enable visual detection of the target sequence (analyte) while eliminating several steps required for pipetting, incubation, and washing out the excess of reactants. In this paper, we present, for the first time, lateral flow tests exploiting oligonucleotide-functionalized and antibody-functionalized carbon nanoparticles (carbon nano-strings, CBNS) as reporters that enable confirmation of the target DNA sequence by hybridization. The CBNS reporters were applied to (a) the detection of PCR products and (b) visual genotyping of single nucleotide polymorphisms in human genomic DNA. Biotinylated PCR product was hybridized with a dA-tailed probe. In one assay configuration, the hybrid is captured at the test zone of the strip by immobilized streptavidin and detected by (dT) 30 -CBNS. In a second configuration, the hybrids are captured from immobilized (dA) strands and detected by antibiotin-CBNS. As low as 2.5 fmol of amplified DNA can be detected. For visual genotyping, allele-specific primers with a 5′ oligo(dA) segment are extended by DNA polymerase with a concomitant incorporation of biotin moieties. Extension products are detected either by (dT) 30 -CBNS or by antibiotin-CBNS. Only three cycles of extension reaction are sufficient for detection. No purification of the PCR products or the extension product is required.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular recognition through hybridization constitutes the cornerstone of nucleic acid detection and quantification. In the last four decades, we have seen a rapid evolution of hybridization methods. For a long time, DNA analysis was based on size separation by electrophoresis followed by transfer to a membrane (Southern transfer), hybridization with radioisotope-labeled probes, and detection by autoradiography. Subsequently, radioisotopes were replaced by enzyme reporters, thus avoiding the health hazards associated with the use and disposal of radioactivity. The invention of polymerase chain reaction (PCR) contributed greatly to the detectability and specificity of nucleic acid tests and enabled detection of minute amounts of DNA/RNA (down to a few copies) in a variety of clinical, environmental, food, and forensic samples. Thus, the sequence of interest is first amplified, exponentially, by PCR and the reaction products are analyzed by electrophoresis or hybridization, the later being advantageous since it allows sequence confirmation. Currently, hybridization assays are all nonradioactive and performed in a heterogeneous or homogeneous format that facilitates automation and high-throughput analysis. Heterogeneous assays involve separation of the hybridized probe from the free probe via immobilization on a solid phase (e.g., microtiter wells or beads). Enzymes (with chemiluminogenic [1], fluorogenic [2], or chromogenic [3] substrates), fluorophores [4], electroactive [5] compounds, and nanoparticles [6] have been used as reporters. Homogeneous hybridization assays are based on fluorescence resonance energy transfer [7–9]. However, all these methods require costly instruments, involve several steps of pipetting, incubation, and washing, and demand highly qualified personnel.
Lateral flow devices were originally designed to perform immunoassays in which the washing steps are eliminated and the presence of the analyte can be observed visually. The pregnancy test is a typical example. Visual detection was achieved by employing strongly colored particles, such as gold [10, 11], polystyrene, carbon black [12, 13], and liposomes [14], as reporters. These unique advantages have, recently, stimulated a growing activity toward the development of lateral flow devices for nucleic acid analysis by employing the same reporters [15–20].
During the last 2 years, carbon nanoparticles were introduced as labels in lateral flow tests for the visual detection of PCR products obtained from various pathogens [18–20]. The upstream and downstream primers were labeled with the hapten digoxigenin and a biotin moiety, respectively. As a consequence, the generated amplification product carried both labels, one at each end. The amplified double-stranded DNA was captured at the test zone of the strip by an immobilized antidigoxigenin antibody and detected by avidin-coated carbon nanoparticles. It is worth noting, however, that this assay configuration simply detects double-labeled DNA without confirmation of the sequence of the amplified product. Any nonspecific amplification product that carries both labels, such as primer dimers, would also be detected by the current configuration.
In the present work, lateral flow tests were developed that enable confirmation of the target DNA sequence by hybridization. In addition to avidin-coated carbon nanoparticles, we have prepared oligonucleotide-conjugated carbon nanoparticles. The functionalized carbon nanoparticles were exploited as reporters for (a) detection of PCR products and (b) visual genotyping of single nucleotide polymorphisms (SNPs) in human genomic DNA. We have employed a newly introduced type of carbon nanoparticles, named carbon black nano-strings (CBNS). CBNS are elongated nanoparticles containing smaller, roughly spherical particles that have been fused together in the production process to form chain-like and branched structures. CBNS have been introduced recently as reporters for the development of an ultrasensitive lateral flow immunoassay [21].
The aforementioned devices were applied (as model assays) to: (a) the detection of PCR-amplified DNA from the invertase gene, that is a maize-specific gene used as a reference gene in the testing for genetically modified organisms [22] and (b) to the genotyping of a SNP in the toll-like receptor 4 (TLR4) gene. Toll-like receptors play a key role in pathogen recognition and the activation of innate immunity. SNPs in the TLR4 gene are associated with airway hyporesponsiveness to inhaled endotoxin [23] and may have an impact on the susceptibility to various infectious and inflammatory diseases.
Materials and methods
Apparatus and reagents
PCR amplification and primer extension reactions were performed in the MJ Research PTC-0150 thermal cycler (Watertown, MA). A digital camera, Kodak DC 120, and the Gel Analyzer software for DNA documentation were purchased from Kodak (New York, NY). An ultrasonic cleaner from Branson Ultrasonics (Danbury, CT) was used for resuspension of the carbon black nano-strings. The TLC applicator, Linomat 5, and the software WinCats were from Camag (Muttenz, Switzerland).
CBNS, 100 and 200 nm in size, were purchased from Maiia Diagnostics (Uppsala, Sweden). Antibiotin antibody, sulfo-SMCC (Sulfosuccinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate) and SATA (N-succinimidyl-S-acetylthioacetate) were from Pierce chemicals (Rockford, IL). Terminal deoxynucleotidyl transferase (TdT) was from MBI Fermentas (Vilnius, Lithuania). Taq and Vent (exo-) DNA polymerase were from New England Biolabs (Beverly, MA). Ultrapure 2′-deoxyribonucleoside 5′-triphosphates (dNTPs) were purchased from Invitrogen (CarlsBad, CA). Biotin-16-dUTP was from Clontech (Mountain View, CA). Immunopore FP nitrocellulose membrane was purchased from Whatman (Florham Park, NJ). The wicking pad, glass-fiber conjugate pad, and absorbent pad were from Schleicher & Schuell (Dassel, Germany). Streptavidin from Streptomyces avidinii was purchased from Roche Diagnostics (Mannheim, Germany). Sephadex G-25 and bovine serum albumin (BSA) were from Sigma (St. Louis, MO). All other common reagents were from Sigma (St. Louis, MO). The primers and probes used in this study were synthesized by Thermo Electron (Ulm, Germany). For the PCR of the IVR gene, the upstream and downstream primer sequences were: 5′-Biotin-CCGCTGTATCACAAGGGCTGGTACC-3′ and 5′-GGAGCCCGTGTAGAGCATGACGATC-3′, respectively. The IVR detection probe was: 5′-AGGTGCAGCCAGTGGAGGAGGTCG-3′. For the PCR of the TLR4 gene [24], the upstream and downstream primer sequences were: 5′-GTAAAACGACGGCCAGTAGTCCATCGTTTGGTTCTGGGAGA-3′ and 5′-CAGGAAACAGCTATGACGCCATTGAAAGCAACTCTGGTGTG-3′, respectively. The allele-specific primers (normal and mutant) for SNP genotyping were: 5′-A24GTGATTTTGGGACAAC and 5′-A24GTGATTTTGGG ACAAT-3′, respectively.
Functionalization of CBNS with antibiotin antibody
An aliquot containing 10 μg of CBNS was diluted in 500 μL of 5 mM borate buffer, pH 8.4, sonicated for 2 min, and mixed with 500 μL of the same buffer containing 4 μg of antibiotin antibody. The mixture was incubated for 1 h at room temperature. CBNS were then blocked for 30 min at ambient temperature with 100 μL of 100 g/L BSA in 50 mM borate buffer, pH 8.4. The mixture was centrifuged for 5 min at 16,000×g and the CBNS were washed four times with 100 μL of 10 g/L BSA, 0.02% NaN3 in 100 mM borate buffer, pH 8.4. The conjugated nano-strings were finally resuspended in 100 μL of the same buffer and sonicated for 5 min. An aliquot of 5 μL was applied to the conjugate pad of the strip.
Coupling of (dT)30 oligonucleotide to CBNS
An amount of 50 μg of CBNS in 500 μL of 5 mM borate buffer, pH 8.4, was sonicated for 2 min and mixed with 500 μL of 10 g/L BSA in the same buffer. The mixture was incubated for 1 h at room temperature. The CBNS were pelleted by centrifugation at 16,000×g for 5 min and washed twice with 100 μL of 10 g/L BSA in phosphate-buffered saline (PBS), pH 7.4. The pellet was finally resuspended in 20 μL PBS, pH 7.4, and sonicated for 5 min before use.
A 10-μL aliquot of albumin-coated CBNS was mixed with 10 μL of 0.1 M phosphate buffer, pH 7.4, and 5 μL of 87.3 μg/μL (1 μmol) of SMCC and the reaction was allowed to proceed for 1 h at room temperature. Then, 50 μL of 10 g/L BSA in 0.1 M phosphate buffer, pH 7.4, were added and the mixture was centrifuged for 5 min at 16,000×g. The pellet was washed twice with the same buffer and resuspended in 10 μL of 0.1 M phosphate buffer, pH 7.4. The conjugate was sonicated for 2 min before use.
The (dT)30 oligonucleotide was derivatized with SATA. The reaction mixture consisted of 2 μL of 300 pmol/μL 5′ amino-modified (dT)30, 2 μL of 6.9 g/L SATA, and 2 μL of 0.15 M NaHCO3, pH 9.1. The mixture was incubated for 1 h at room temperature. The volume was adjusted to 20 μL with water, and the derivatized oligo was purified twice by size-exclusion chromatography using Sephadex G-25 spin-pure columns.
Finally, SATA-(dT)30 reacted with SMCC-CBNS. The coupling reaction mixture consisted of 10 μL SMCC-CBNS, 20 μL of SATA-(dT)30, 6 μL of 5× conjugation buffer (0.5 M phosphate buffer, pH 7.4, 25 mM EDTA), and 7.2 μL 0.5 M hydroxylamine in 1× conjugation buffer. The mixture was incubated for 1 h at room temperature. Then, 50 μL of 10 g/L BSA in 100 mM borate buffer, pH 8.4, were added and the mixture was centrifuged for 5 min at 16,000×g. The pellet was washed twice with 100 μL of the same buffer and resuspended in 50 μL buffer.
Tailing of oligonucleotide probes with dATP or dTTP
The oligonucleotide probe (IVR probe), specific for the invertase (IVR) gene, and the (dT)30 oligonucleotide were tailed at the 3′ end with dATP or dTTP, respectively, using terminal deoxynucleotidyl transferase. The tailing reaction was carried out in a final volume of 20 μL containing 0.2 M potassium cacodylate, 25 mM Tris (pH 7.2), 1 mM CoCl2, 0.1 ml/L Triton X-100, 2 mM dATP (or 5 mM dTTP), 400 pmol IVR probe or 1,000 pmol (dT)30, and 30 U transferase. The mixture was incubated for 1 h at 37 °C, and the reaction was stopped by adding 2 μL of 0.5 M EDTA (pH 8.0). Methylmaleimide was also added at a final concentration of 2.5 mM to the (dA)-tailed probe.
Preparation of the lateral flow strip
The dry-reagent strip (4 × 70 mm) consisted of an immersion pad, a conjugate pad, a laminated membrane, and an absorbent pad assembled on a plastic adhesive backing that provides rigidity. The four parts were positioned in such a way that their ends overlapped in order to ensure continuous flow (by capillary action) of the developing solution from the wicking pad up to the absorbent pad. Two types of strips were constructed. For the first type, streptavidin (27 pmol) was immobilized by physical adsorption on the test zone of the strip. Similarly, poly(dA) (2.4 pmol) was immobilized on the control zone of the strip. For the construction of the second type of biosensor, 10 pmol of polydT strands were immobilized on the test zone of the biosensor, whereas biotinylated BSA (50 ng) was immobilized at the control zone of the strip. A 5-μL aliquot of the (dT)30-CBNS and antibiotin-CBNS were placed on the conjugate pad, respectively.
PCR
PCR of a 225-bp segment of the maize-specific invertase (IVR) gene from genomic DNA standard (69407 GMO Genomic DNA Standard Set for Maize NK603, GA21 and CBH-351) was carried out in a total volume of 50 μL containing 2 U of HotStar Taq DNA polymerase, 1× PCR buffer, 200 μM of each dNTP, and 0.5 μM of each primer. The PCR conditions were as follows: 5 min at 95 °C followed by 35 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 40 s. At the end of the cycling, the mixture was held at 72 °C for 10 min. The PCR products were confirmed by 1.7% agarose gel electrophoresis and ethidium bromide staining and quantified by densitometry.
Visual detection of PCR products by lateral flow hybridization test using CBNS as reporters
Aliquots of PCR product were mixed with 1 μL of 0.9 M NaCl and 1 pmol of dATP-tailed IVR probe in a total volume of 10 μL. The mixture was heated at 95 °C for 2 min and then incubated at 37 °C for 5 min. This step was performed in the thermocycler. A 5-μL aliquot of the mixture was applied to the conjugate pad of the strip. The strip was then immersed into the developing solution containing 10 mL/L glycerol, 10 g/L SDS and 10 mL/L Tween-20, 10 mg/mL BSA in PBS, pH 7.4. A gray/black line at the test zone of the sensor denotes the presence of the amplified product. A second black line is formed at the control zone to confirm the proper function of the sensor. The detection is completed within 10–15 min.
Visual genotyping of single nucleotide polymorphisms by lateral flow hybridization test using CBNS as reporters
A 634-bp segment flanking the C1196T polymorphic site of the TLR4 gene was PCR amplified from human genomic DNA. Amplification was carried out in a total volume of 50 μL containing 25 μL of master mix (0.05 U/μL Hotstar DNA polymerase, 1× PCR buffer, 1.5 mM MgCl2, and 200 μΜ dNTPs), 0.4 μΜ upstream and downstream primers, and 60–120 ng of genomic DNA. The thermal cycling protocol included the following steps: 95 °C for 15 min followed by 35 cycles of 95 °C for 1 min, 65 °C for 1 min, 72 °C for 1 min, and a final extension step at 72 °C for 8 min. PCR products were stored at 4 °C.
PCR-amplified DNA was subjected to two separate primer extension reactions, one with the normal (N) primer and the other with the mutant (M) primer. Both primers contained a (dA)24 tail at the 5′ end. The reaction was carried out in a total volume of 20 μL containing 20 mM Tris–HCl, pH 8.8, 10 mM (NH4)2SO4, 10 mM KCl, 0.1% Triton X-100, 0.5 U Vent (exo-) DNA polymerase, 1–2 μL of amplified DNA, 1 pmol of the appropriate primer, 2 mM MgSO4, 2.5 μM each dATP, dCTP, dGTP, 1.25 μΜ dTTP, and 1.25 μΜ biotin-dUTP. The reactions were performed in the thermal cycler as follows: 95 °C for 5 min and three cycles of 95 °C for 15 s, 55 °C for 10 s, and 72 °C for 15 s. The products were subjected to a final denaturation step at 95 °C for 5 min and placed immediately on ice. An aliquot of the denatured product (3–5 μL) was applied onto the conjugate pad next to the deposited conjugated nano-strings. The wicking pad was then dipped into a microcentrifuge tube containing 200 μL of 10 mL/L glycerol, 10 g/L SDS and 10 mL/L Tween-20, 10 g/L BSA in PBS, pH 7.4. The products were detected visually in about 10–15 min.
Results and discussion
Functionalization of carbon nano-strings with antibody or oligonucleotide
We prepared two types of functionalized CBNS to be used as reporters in lateral flow hybridization assays. (a) CBNS conjugated with oligonucleotide ((dT) 30 -CBNS). The conjugation strategy is outlined in Fig. 1. A (dT)30 oligonucleotide carrying a –NH2 group at the 5′ end was derivatized with SATA, thereby introducing a protected sulfhydryl group at the 5′ end. The nano-strings were coated (by adsorption) with albumin to create a surface that is rich in primary amino groups. Then, the –NH2 groups reacted with the heterobifunctional linker sulfo-SMCC, thus creating a surface with maleimide groups. The derivatized CBNS and (dT)30 were mixed and hydroxylamine was added in order to deprotect the –SH groups and initiate the conjugation of the nano-strings with the (dT)30. (b) CBNS coated with antibiotin antibody (antibiotin-CBNS) was prepared by adsorption of the antibody onto the surface of the nano-strings and subsequent blocking of the uncovered sites with bovine serum albumin.

Outline of the strategy used for the conjugation of carbon black nano-strings (CBNS) with the oligonucleotides. (a) Derivatization of the 5′ NH2-oligonucleotide with SATA. (b) Reaction of albumin-coated CBNS with sulfo-SMCC and subsequent coupling to the derivatized oligonucleotide
Both the (dT) 30 -CBNS and the antibiotin-CBNS reporters were prepared by using the CBNS 100 and 200 nm in size.
Lateral flow hybridization tests for visual detection of PCR products
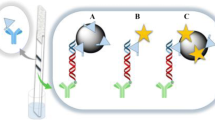
Figure 2 illustrates the design of the strip and the principles of the lateral flow hybridization tests that exploit functionalized CBNS as reporters for visual detection of PCR products. We developed two assay configurations. The first configuration (Fig. 2B) uses (dT) 30 -CBNS reporters. The amplification product (target), which is biotinylated at the 5′ end through the upstream PCR primer, was denatured and hybridized (for 5 min) in the thermal cycler with an oligonucleotide probe that carried one segment complementary to the target and an oligo(dA) tail at the 3′ end. The hybrid is applied to the conjugate pad of the strip which is then immersed in the developing buffer. As the buffer flows along the strip, by capillary action, the (dT) 30 -CBNS, which is also deposited on the conjugate pad, is resuspended and hybridizes to the oligo(dA) tail of the probe. The hybrid is captured from immobilized streptavidin at the test zone of the strip. The accumulation of CBNS at the test zone gives a black line to signify the presence of PCR product. In the absence of amplified DNA the oligo(dA)/dT30 hybrids are formed but are not captured on the strip. The excess of (dT) 30 -CBNS is captured from the immobilized oligo(dA) strands at the control zone of the strip thus forming always a black line that confirms the proper function of the test.

(A) Design of the strip. The arrow shows the direction of the flow. (B) and (C) Assay configurations of the lateral flow hybridization tests for visual detection of PCR products by exploiting (dT) 30 -CBNS or antibiotin-CBNS reporters, respectively. IP immersion pad, CP conjugate pad, M membrane, AP absorbing pad, TZ test zone, CZ control zone, SA streptavidin, B biotin, BSA bovine serum albumin
The second assay configuration (Fig. 2C) exploits antibiotin-CBNS as reporters. Here, the test zone of the strip contains immobilized oligo(dA) strands. The antibiotin-CBNS binds to the biotin moiety of the PCR product and the hybrids are captured at the test zone through dA/dT strand interaction. The excess of antibiotin-CBNS binds to biotinylated albumin that is immobilized at the control zone of the strip, thus generating a second black line.
We optimized the amount of (dT)30 oligo in the coupling to CBNS reaction. Aliquots containing 10 to 1,000 pmol oligo were derivatized with SATA and mixed with a constant amount of 25 μg of maleimide-derivatized albumin-coated CBNS. The experiments were performed by using CBNS of 100 and 200 nm in size. As a model target, we used a 5′ biotinylated single-stranded DNA (10 fmol) that carried a poly(dA) tail at the 3′ end. The results are presented in Fig. 3. We observe that the intensity of the test zone increases with the (dT)30 load up to 300 pmol for the 100 nm CBNS and up to 1,000 pmol of oligo for the 200 nm CBNS. We decided to carry out all the following studies with CBNS (100 or 200 nm) that are conjugated with 600 pmol of (dT)30.

Studies of (dT)30 to CBNS conjugation. (A) Effect of the amount of (dT)30 in the conjugation reaction using 100 nm CBNS. (B) Effect of the amount of (dT)30 in the conjugation reaction using 200 nm CBNS. (C) Comparison of (dT) 30 -CBNS reporters with the poly(dT)-CBNS reporters (CBNS of 100 nm). TZ test zone, CZ control zone
We then investigated whether the tailing of (dT)30 with dTTP improves the detectability of the lateral flow test due to the fact that longer dT strands attached on the surface of the nano-strings might allow more effective hybridization. An amount of 10 fmol of the target DNA was tested, in parallel, by using strips containing CBNS (100 nm) coupled to 300 pmol of either (dT)30 or poly(dT) strands. The results (Fig. 3) show that the use of long poly(dT) strands has no effect on the intensity of the zone.
To assess the ability of the (dT) 30 -CBNS based strip tests and the antibiotin-CBNS strip tests to detect double-stranded DNA, we applied various amounts of PCR products (225 bp amplified DNA) ranging from 0 to 200 fmol per strip. Both assay configurations were assessed in parallel. We observe (Fig. 4) that as low as 2.5 fmol of PCR product (0.37 ng) can be detected by the two types of strip tests. Furthermore, the intensity of the test zone increases with the amount of PCR product. At the level of 200 fmol, the intensity drops slightly. The effect of the size of carbon nano-strings on the detectability of the strip tests was also studied, using CBNS of 100 or 200 nm in size. The 200 nm CBNS gave slightly higher zone intensity.

Performance of the lateral flow hybridization tests for visual detection of PCR products. The amount of PCR product, in fmoles, is shown above the strips. The results were obtained from lateral flow hybridization tests based on: (A) (dT) 30 -CBNS reporters, with 100 nm CBNS; (B) (dT) 30 -CBNS reporters, with 200 nm CBNS; (C) antibiotin-CBNS reporters, with 100 nm CBNS; and (D) antibiotin-CBNS reporters, with 200 nm CBNS. TZ test zone, CZ control zone
To assess the reproducibility of both assay configurations, samples containing 8 or 50 fmol of PCR product were tested in triplicate using strips based on (dT) 30 -CBNS and antibiotin-CBNS reporters. CBNS of 100 and 200 nm in size were employed. The results are presented in Fig. 5. The strips were scanned with a simple desktop scanner (Hewlett-Packard ScanJet 3400C) and the color intensity of the test zone was determined by densitometry. The (dT) 30 -CBNS based assay configuration gave CVs, for the 100 nm CBNS, of 3.3% (8 fmol target) and 7.3% (50 fmol target). The CVs obtained for the 200 nm CBNS reporter were 3.4% (8 fmol) and 4.2% (50 fmol). The antibiotin-CBNS based assay configuration gave CVs, for the 100 nm CBNS, of 6.4% (8 fmol) and 1.2% (50 fmol). The CVs obtained for the 200 nm CBNS reporter were 5.4% (8 fmol) and 6.0% (50 fmol).

Reproducibility study. Samples containing 8 or 50 fmol of PCR product were tested in triplicate using lateral flow tests based on (dT) 30 -CBNS and antibiotin-CBNS reporters. (A) (dT) 30 -CBNS reporters, with 100 nm CBNS. (B) (dT) 30 -CBNS reporters, with 200 nm CBNS. (C) Antibiotin-CBNS reporters, with 100 nm CBNS. (D) Antibiotin-CBNS reporters, with 200 nm CBNS. TZ test zone, CZ control zone
Lateral flow tests for visual genotyping of single nucleotide polymorphisms
Figure 6 illustrates the principles of the lateral flow hybridization tests that exploit functionalized CBNS as reporters for visual genotyping of SNPs. PCR-amplified DNA is subjected to two separate primer extension reactions, each with an allele-specific primer. Both primers contain a (dA)24 segment at the 5′ end and differ only in one base at the 3′ end. Due to the specificity of DNA polymerase, a primer is extended only if the perfectly complementary to the target sequence (Fig. 6A). Biotin-dUTP is incorporated into the extended fragment. We developed two assay configurations for visual detection of the extension product by a lateral flow hybridization test. The first configuration (Fig. 6B) exploits (dT) 30 -CBNS reporters. The extension product hybridizes with the (dT)30 strands on the CBNS and the hybrid is captured by immobilized streptavidin at the test zone of the strip, thus creating a black line. Excess of (dT) 30 -CBNS is captured by immobilized poly(dA) strands at the control zone of the strip. The second configuration (Fig. 6C) employs antibiotin-CBNS reporters. Antibiotin-CBNS binds to the biotins that are incorporated in the extension and the complex is captured at the test zone through hybridization with immobilized poly(dA) strands. Excess of antibiotin-CBNS is captured by immobilized biotinylated albumin at the control zone of the strip.

(A) The allele-specific primer extension reaction. (B) and (C) Assay configurations of the lateral flow hybridization tests for visual genotyping of single nucleotide polymorphisms by exploiting (dT) 30 -CBNS or antibiotin-CBNS reporters, respectively. TZ test zone, SA streptavidin, B biotin, BSA bovine serum albumin. (D) Visual genotyping of a single nucleotide polymorphism in three samples of human genomic DNA (a normal, a homozygote for the mutation and a heterozygote) by lateral flow hybridization tests based on (dT) 30 -CBNS reporters and antibiotin-CBNS reporters. The genotype is shown above the strips
The genotype is obtained by the results of two strips corresponding to the extension reaction of the normal primer (N) or mutant primer (M). Thus a normal genotype (N/N genotype) gives a black line only for the extension reaction of the N-primer. A homozygote for the mutation (M/M genotype) gives a black line only for the extension of the M-primer. A heterozygote (N/M genotype) gives both strips with positive results.
As a model, we have used the toll-like receptor 4 (TLR4) gene for genotyping the C1196T polymorphism in 15 samples of human genomic DNA. Genomic DNA samples were obtained from previous extensive studies based on informed consent [24, 25]. All genotyping experiments were carried out using two configurations of lateral flow tests based on either (dT) 30 -CBNS or antibiotin-CBNS (CBNS of 200 nm) as reporters. The results are presented in Fig. 6(D) and Figure S1 (see Electronic Supplementary Material). Both assay configurations give identical genotypes. The results are fully concordant with sequencing data.
Conclusions
We developed and evaluated lateral flow hybridization tests based on CBNS that are conjugated either to an oligonucleotide or an antibody, for the detection of double-stranded DNA (PCR products) and genotyping of single nucleotide polymorphisms. Exponential amplification of DNA or RNA (usually by PCR) constitutes an essential step of modern methods of nucleic acid analysis. Considerable effort has been put in the development of methods for detection of amplified products. Electrophoresis combined with ethidium bromide staining is still the most common approach. However, it is time-consuming and offers size not sequence confirmation. Homogeneous or heterogeneous hybridization assays are automatable and offer sequence confirmation but require costly equipment and reagents. On the contrary, the proposed lateral flow device is extremely simple, enables visual detection of the PCR product and sequence confirmation through hybridization without the use of instruments for detection. The only instrument required is a simple (inexpensive) thermal cycler, which is considered a common equipment for every laboratory involved in nucleic acid analysis. The equipment required for manufacturing the lateral flow device (a sprayer and a cutter) are not utilized by the analyst. The PCR step takes about 1 h, and the detection step by the lateral flow device is complete within 15 min.
The high detectability of the strip test facilitates SNP genotyping since it allows visual genotyping after only three cycles of primer extension reaction (7 min). The extension products are detected directly by the strip without prior purification of the DNA after the PCR step or after the primer extension reaction step. The assays are simple, rapid (visual detection within minutes), inexpensive, and require no specialized instruments. The lateral flow format eliminates multiple incubation and washing steps and minimizes the need for qualified personnel. The preparation of CBNS conjugates needs about 2 h for the antibody conjugates and 4 h for the oligonucleotide conjugates. Furthermore, the CBNS and CBNS conjugates show great stability in solutions with high ionic strength. Under the current conditions, one vial containing 10 mg of CBNS is sufficient for the preparation of (dT) 30 -CBNS for 4,000 strips and antibiotin-CBNS for 20,000 strips.
References
Elenis DS, Ioannou PC, Christopoulos TK (2007) Anal Chem 79:9433–9440
Bortolin S, Christopoulos TK, Verhaegen M (1996) Anal Chem 68:834–840
Su X, Teh HF, Lieu X, Gao Z (2007) Anal Chem 79:7192–7197
Kalogianni DP, Elenis DS, Christopoulos TK, Ioannou PC (2007) Anal Chem 79:6655–6661
Chen J, Zhang J, Wang K, Lin X, Huang L, Chen G (2008) Anal Chem 80:8028–8034
Li G, Li X, Wan J, Zhang S (2009) Biosens Bioelectron 24:3281–3287
Duan X, Liu L, Wang S (2009) Biosens Bioelectron 24:2095–2099
Jothikumar P, Hill V, Narayanan J (2009) Biotechniques 46:519–524
Ahmad AI, Ghasemi JB (2007) Anal Bioanal Chem 387:2737–2743
Tippkötter N, Stückmann H, Kroll S, Winkelmann G, Noack U, Scheper T, Ulber R (2009) Anal Bioanal Chem 394:863–869
Zhang MZ, Wang MZ, Chen ZL, Fang JH, Fang MM, Liu J, Yu XP (2009) Anal Bioanal Chem 395:2591–2599
Blazková M, Rauch P, Fukal L (2010) Biosens Bioelectron 25:2122–2128
Posthuma-Trumpie GA, Korf J, van Amerongen A (2008) Anal Bioanal Chem 392:1215–1223
Wen HW, Borejsza-Wysocki W, DeCory TR, Durst RA (2005) Anal Bioanal Chem 382:1217–1226
Kalogianni DP, Bravou V, Christopoulos TK, Ioannou PC, Zoumbos NC (2007) Nucleic Acids Res 35:e23
Kalogianni DP, Goura S, Aletras AJ, Christopoulos TK, Chanos MG, Christofidou M, Skoutelis A, Ioannou PC, Panagiotopoulos E (2007) Anal Biochem 361:169–175
Edwards KA, Baeumner AJ (2006) Anal Bioanal Chem 386:1335–1343
Mens PF, van Amerongen A, Sawa P, Kager PA, Schallig HD (2008) Diagn Microbiol Infect Dis 61:421–427
Noguera P, Posthuma-Trumpie GA, van Tuil M, van der Wal FJ, de Boer A, Moers AP, van Amerongen A (2011) Anal Bioanal Chem 399:831–838
Blažková M, Javůrková B, Fukal L, Rauch P (2011) Biosens Bioelectron 26:2828–2834
Lönnberg M, Drevin M, Carlsson J (2008) J Immunol Methods 339:236–244
Elenis DS, Kalogianni DP, Glynou K, Ioannou PC, Christopoulos TK (2008) Anal Bioanal Chem 392:347–354
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA (2000) Nat Genet 25:187–191
Iliadi AC, Ioannou PC, Traeger-Synodinos J, Kanavakis E, Christopoulos TK (2008) Anal Biochem 376:235–241
Iliadi A, Makrythanasis P, Tzetis M, Tsipi M, Traeger-Synodinos J, Ioannou PC, Rapti A, Kanavakis E, Christopoulos TK (2009) Genet Test Mol Biomarkers 13:849–853
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 169 kb)
Rights and permissions
About this article
Cite this article
Kalogianni, D.P., Boutsika, L.M., Kouremenou, P.G. et al. Carbon nano-strings as reporters in lateral flow devices for DNA sensing by hybridization. Anal Bioanal Chem 400, 1145–1152 (2011). https://doi.org/10.1007/s00216-011-4845-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4845-2